Original link: https://kaopubear.top/blog/2022-06-26-tcga-pancan-cin-signature/
write in front
On June 15, 2022, Nature published two chromosomal instability pan-cancer studies online. One is titled: A pan-cancer compendium of chromosomal instability ; the other is titled: Signatures of copy number alterations in human cancer .
Just like the news we often say “the fewer words the bigger the story”, scientific literature has a potentially established title law, usually the more important and more cutting-edge articles have shorter titles , such as the DNA double helix structure published in Nature in 1953 Molecular Structure of Nucleic Acids.
In many directions of oncology research, today we can only achieve publication by increasing restrictions and attributes. So now that you see research within 10 words, you might as well take a second look. This is in the direction that I am interested in. Nature has made two articles online at a time, which is not bad. Here, let’s talk about the first one first, and we’ll talk about the rest when we have a chance.
Genomic (chromosomal) instability has been known as one of the hallmarks of tumors for 20 years. Whether it is a single cancer or a pan-cancer, TCGA/PCAWG has published many articles before and after, and now these data can also be published in Nature In the magazine, and you can also call it a pan-cancer species chromosomal instability compendium (compendium), there must be something.
And another reason why I really want to take a closer look at this article is its turbulent publication experience. The submission time of the article is July 31, 2020, the acceptance time of Nature is April 21, 2022, and the official online time is June 15, 2022. A submission is close to two years of revision time. For a paper without a wet test, the pull between it and the review is not too long.
The result of four rounds of revision is that the article has a total of 68 figures, 60 pages of attachments, and 90 pages of Peer Review File . Today, we will study this literature together, and try to talk about the possible twists and turns behind its publication and the lessons we can learn.
maybe learn
The following points are some of the literature learning points I have refined after reading, and you can use them as a reference when reading the original text.
- Numerous core concepts, indicators and classic literature related to chromosomal instability
- Extensive publicly available public data related to chromosomal instability
- How can new concepts based on bioinformatics methods be validated?
- How scientific articles based on bioinformatics can be related to clinical research
- What are the challenges of publishing CNS journals based on bioinformatics using public data?
About chromosomal instability
Chromosomal instability (CIN, chromosomal instability) has very complex effects on tumors, including loss or amplification of driver genes, rearrangement of large fragments, extrachromosomal DNA (ecDNA), micronucleus formation, and activation of innate immune signals, etc. . This leads to its association with tumor stage, metastasis, poor prognosis and drug resistance.
The causes of CIN are also diverse, including mitotic errors, replication stress, homologous recombination defects (HRD), telomere abnormalities, and the breakage-fusion-bridge cycle, among others.
Due to the diversity of these causes and consequences, in the current study, CIN is usually viewed as a composite event.
Measurement descriptions of CIN, typically, either classify tumors as high or low CIN, or are limited to a single etiology such as HRD, have or are limited to specific genomic features such as chromosomal arm changes, or Only specific cancer types can be quantified.
There is currently no systematic framework to comprehensively describe the diversity, extent, and origin of pan-cancer CIN, nor to define the relationship between different types of CIN within tumors and clinical phenotypes. (Well, the two articles mentioned at the beginning of the article are both answering this question).
Core research ideas and methods
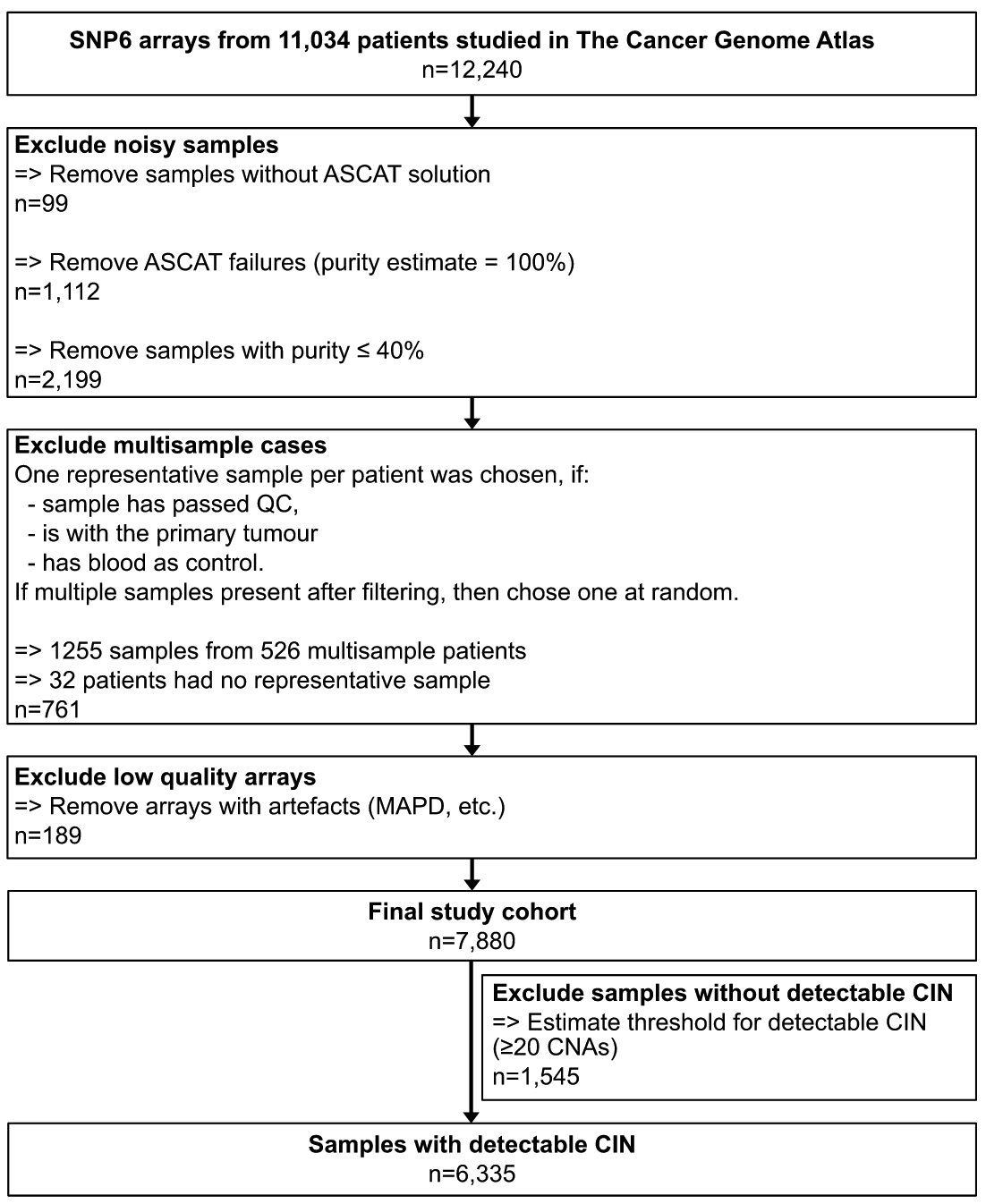
research sample
To address the issues mentioned above, the authors first obtained 7880 high-quality absolute copy number maps for 33 tumor types using SNP6 array data from The Cancer Genome Atlas (TCGA). The software used and the screening process are shown in the figure below.
Feature selection
Using the framework of an ovarian cancer CIN study published by his team in Nat. Genet. in 2018, the authors calculated five copy number signatures that have previously been shown to be valid on 6,335 genome-wide copy number results, which can represent copy number changes mode (potential reasons for different CINs).
These five characteristics include:
- Copy number changes between adjacent chromosome segments
- Fragment length
- Breakpoints per 10Mb
- Number of breakpoints per chromosome arm
- Chain lengths of copy number oscillatory states
If you don’t understand, you can see the detailed description of the original method or the schematic diagram below.

The authors also give some explanations in the attached method as to why some of the more understandable indicators were not taken into account.
For example, absolute copy numbers are excluded because multiple copy number change events may occur at the same genomic location, so the absolute copy number of the segment may vary widely depending on the order of the various mutation events.
For example, a single-copy loss results in a copy number of 3 in the context of whole genome duplication WGD, but 1 in the context of non-whole genome duplication, although they may be caused by the same mutational process that caused the deletion, there will be two different characteristics.
Because chromosomal dorsality can have specific effects on many biological properties, not using absolute values as features can correct for the effects of ploidy and avoid redundant features that encode the same cause in different ploidy backgrounds. (One more thing, in fact, this feature is used in another Nature article published at the same time mentioned at the beginning of the article)
Loss of heterozygosity (LOH) status is not considered because most LOH events are so-called copy loss events, which can be detected by copy number changes. In the dataset used in the article, they found that the median length of LOH events in the entire TCGA was 3.6Mb, which indicates that most events can be detected by SNP6 technology or WGS. But as scientific research increasingly uses shallow WGS and single-cell sequencing, they do not yet provide accurate allele-specific resolution, so specialized LOH signatures may limit the application of this method. (One more thing, in fact, this feature is also used in another Nature article published at the same time mentioned at the beginning of the article)
Seeing this, I have a question: whether these indicators based on sequencing data can be used to describe and distinguish common CIN-related events in biology.
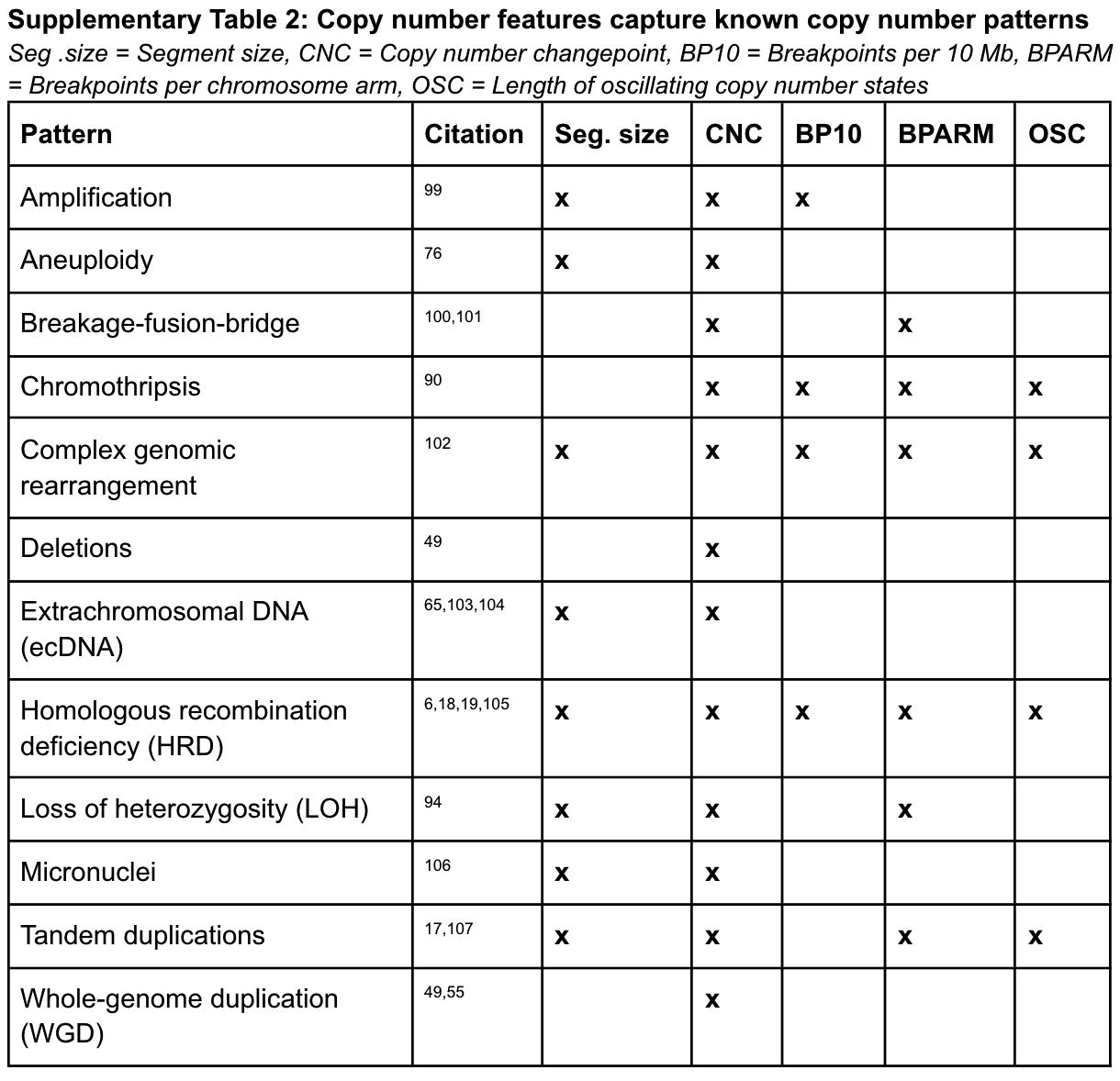
As shown in the figure below, here the authors also list which known copy number patterns can be captured by the above five specific copy number signatures after thorough research. And it is these that prompted the authors to choose these five essential features to mimic known patterns and potentially differentiate their mutational processes.
Signature build
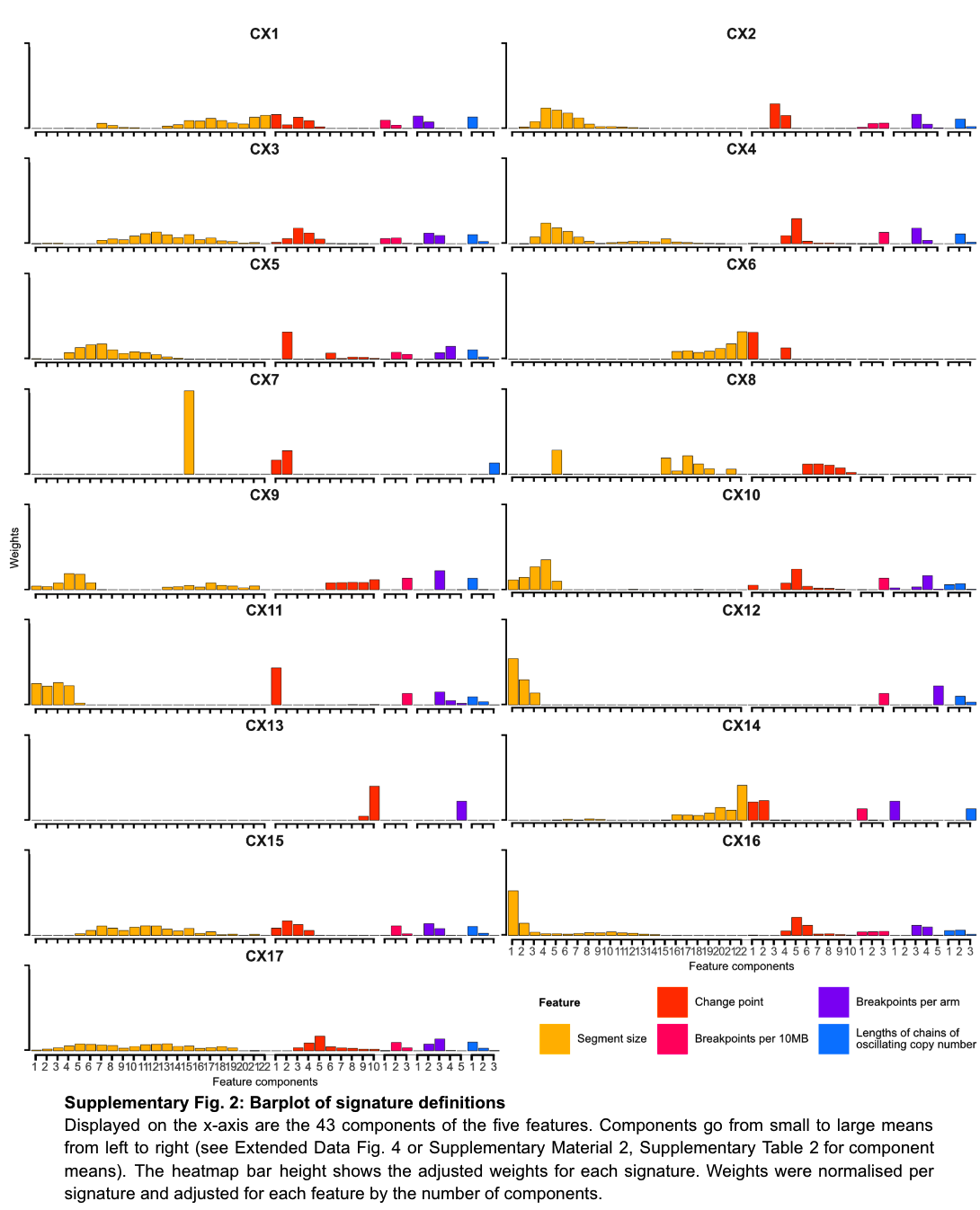
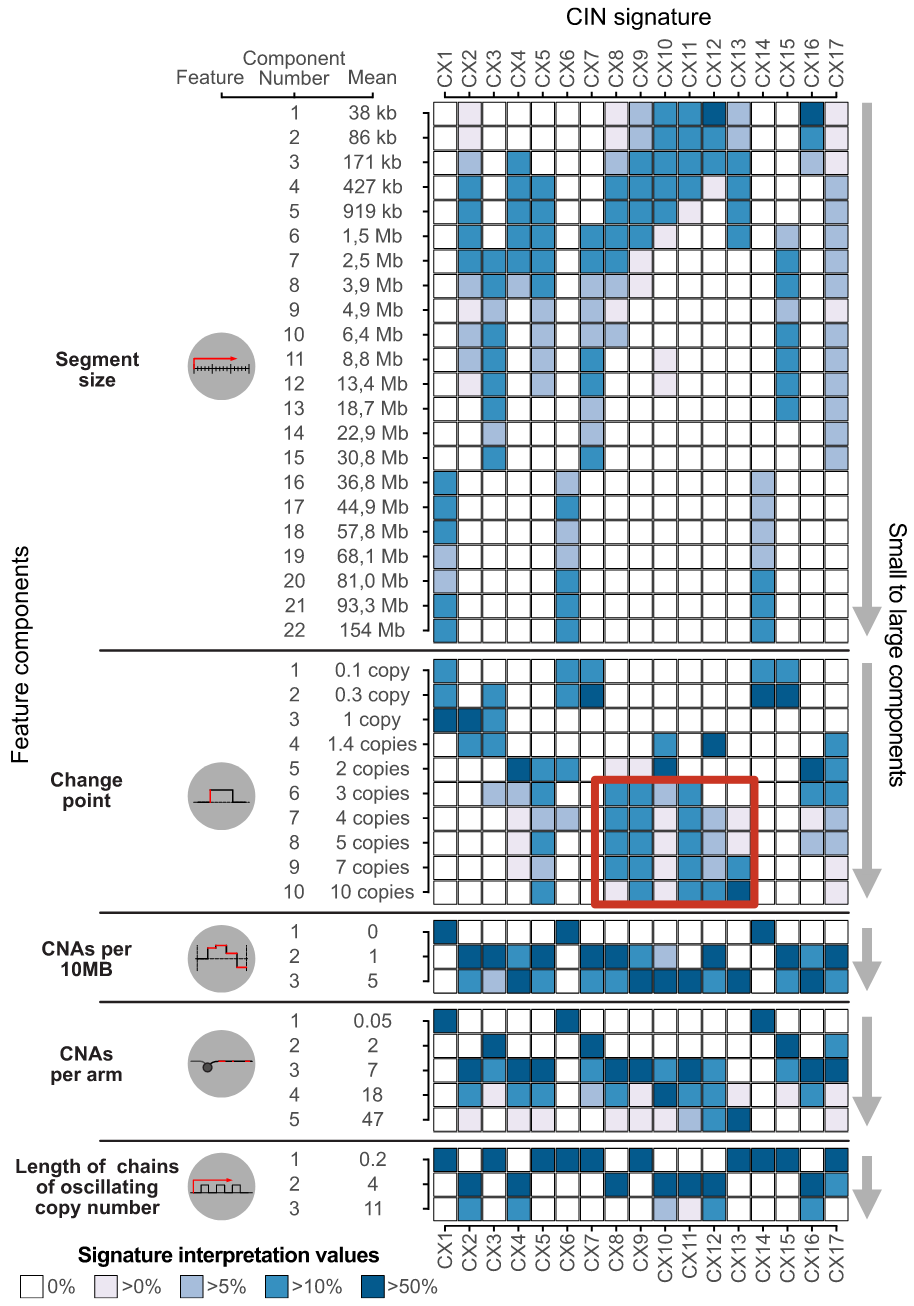
With these 5 features, the authors applied a mixture model to define the different components of the feature distribution within each cohort, and ultimately identified a total of 43 mixture components across the 5 features. The fragment length contains 22 components, and the copy number change contains 10 components, as shown in the following figure.
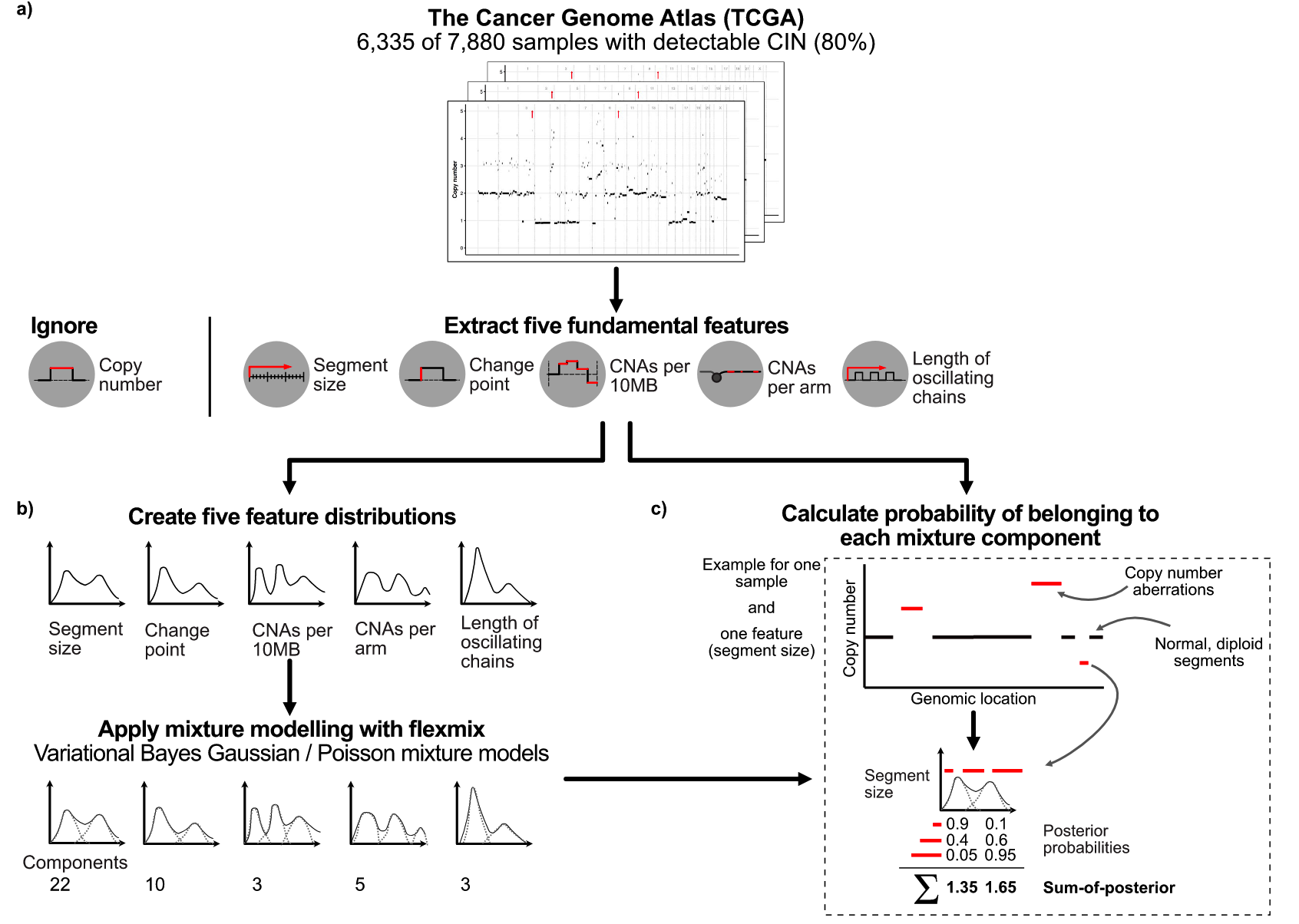
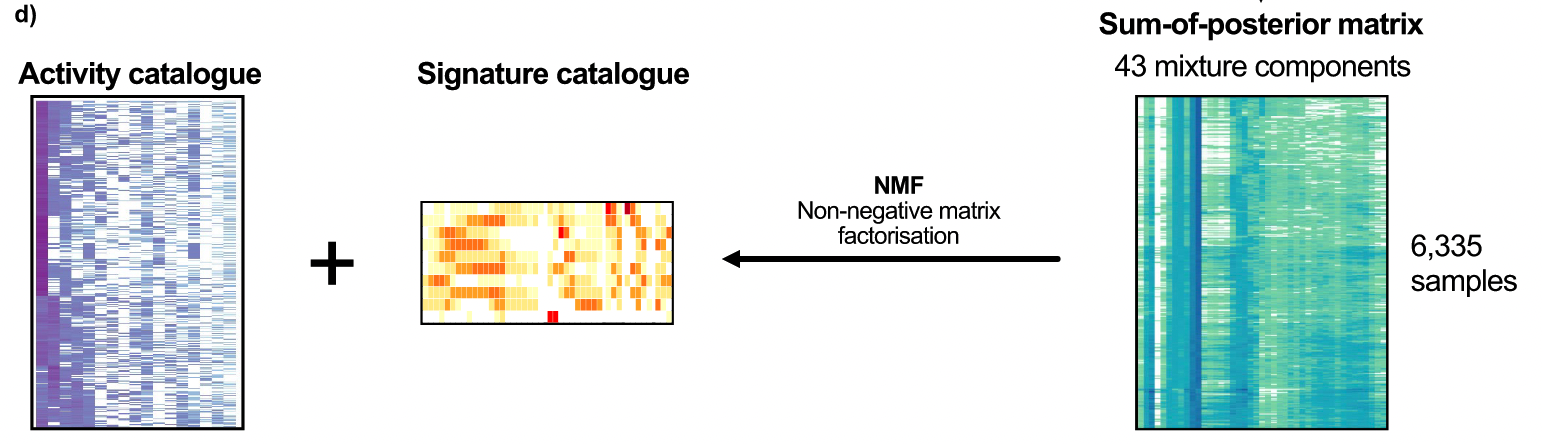
In theory, these 43 components represent the basic building blocks that define the CIN process. These mixed components can then be used to encode each tumor genome and probabilistically assign copy number events to these components, forming a 6,335×43 matrix. Then, non-negative matrix factorization was applied to identify copy number signatures.
main conclusion
17 copy number signatures
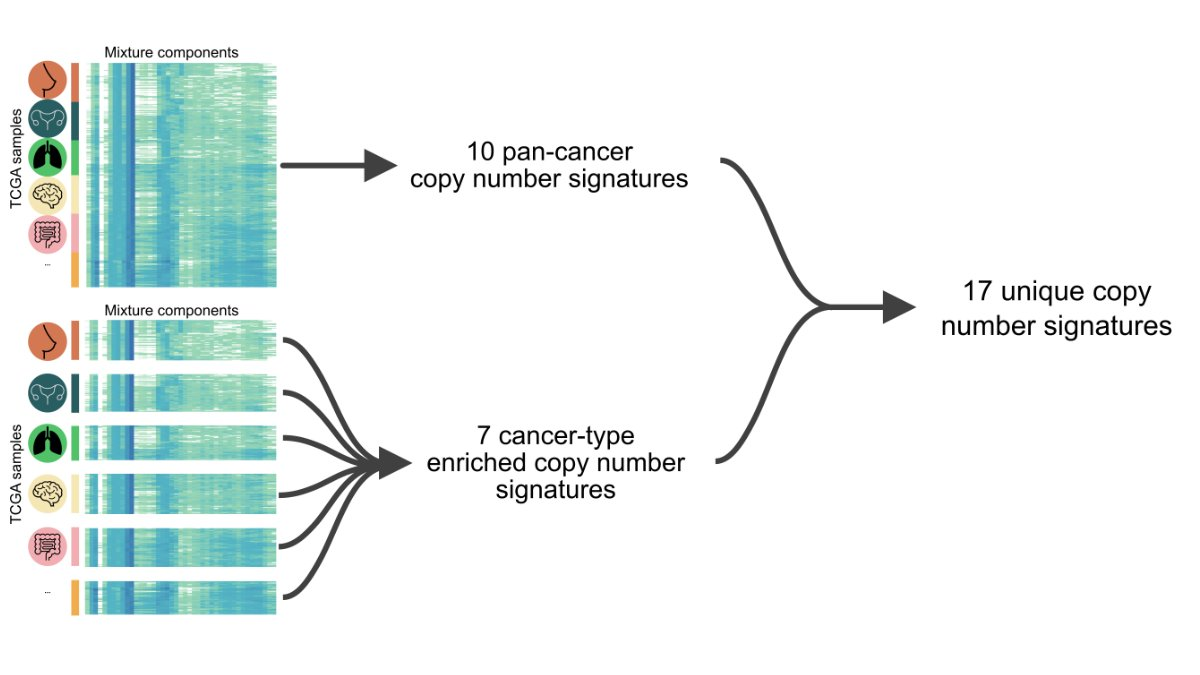
The authors first discovered 10 pan-cancer copy number signatures using the full matrix. A subset of the matrix, representing a single cancer type with at least 100 samples, was then analyzed, first finding 128 cancer species-specific signatures.
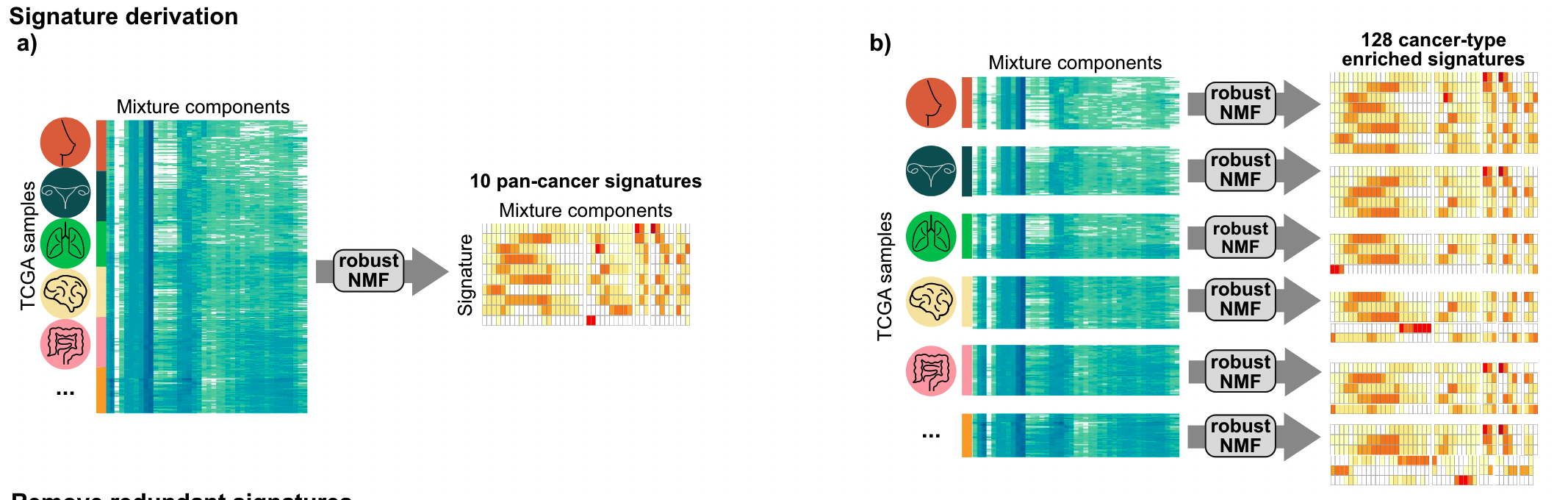
Then a three-step filtering is performed to avoid signature redundancy.
- Delete all cancer species-specific copy number signatures with a cosine similarity greater than 0.74 to any pan-cancer signature
- Select a representative signature from cancer species-specific copy number signatures with a cosine similarity greater than 0.74
- Deletion of cancer species-specific copy number signatures that constitute the pan-cancer signature portfolio
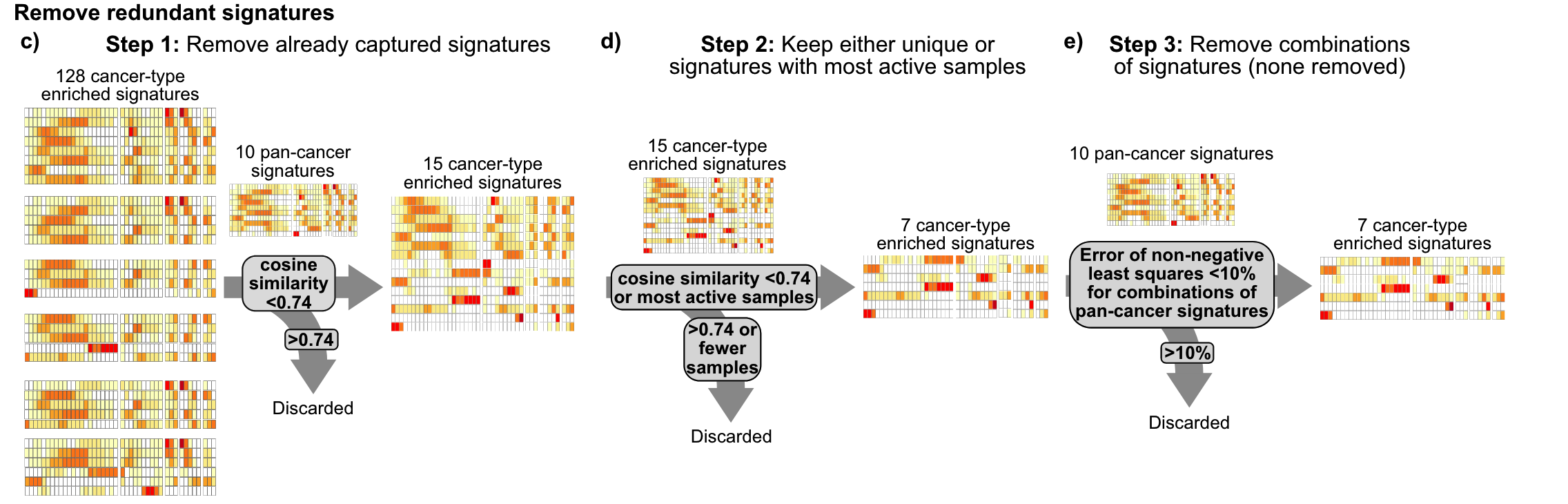
Finally, 17 copy number signatures (10 pan-cancer and 7 cancer species-specific) were found. In the figure below, you can see the 43 components of different copy number features such as fragment length and number of breakpoints in these 17 signatures. Weights.
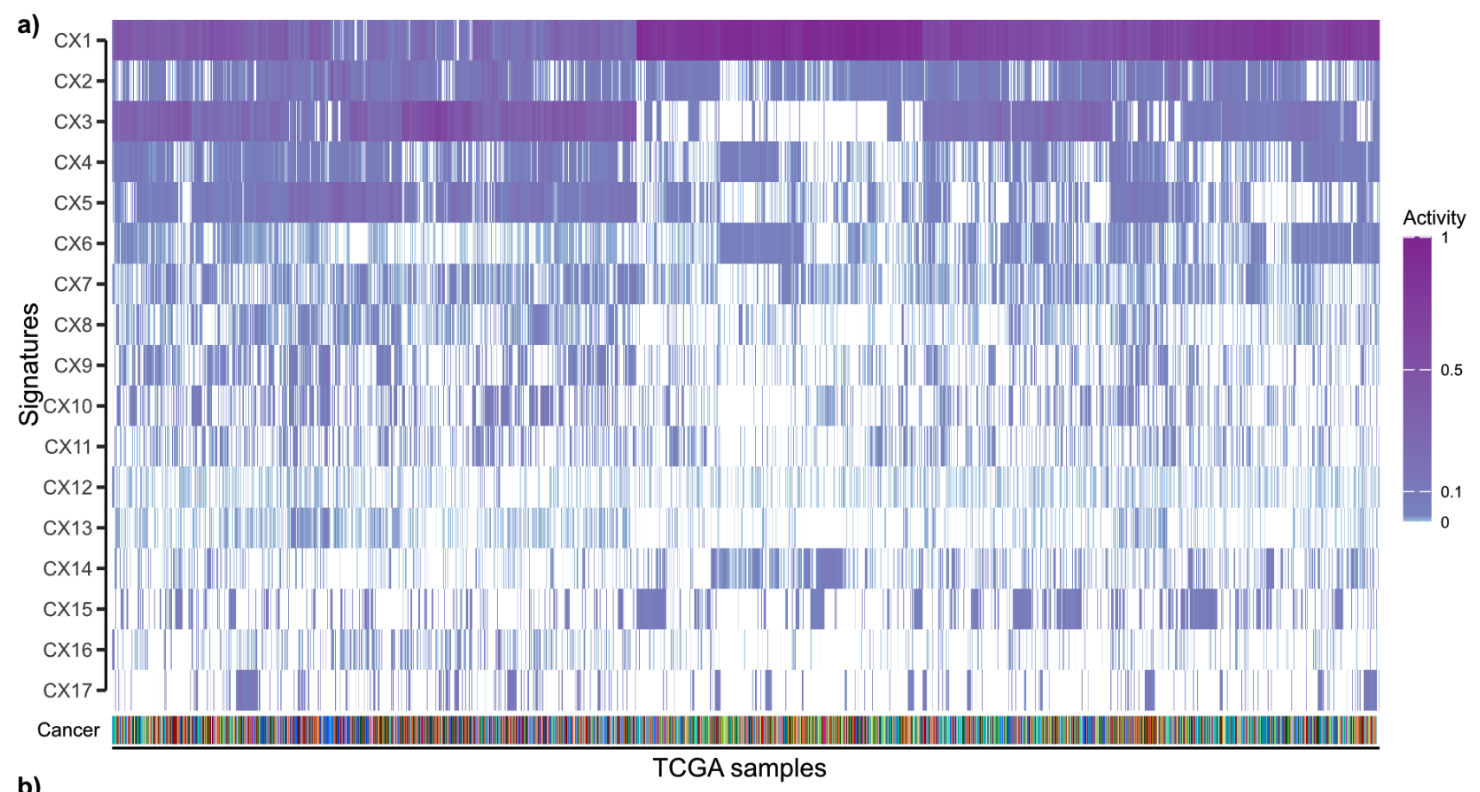
Using linear combination decomposition to calculate their activity, we can finally get a pan-cancer 17 copy number signature and their sample activity in 33 cancer types.
Infer the causes of different signatures
The highlight of this article is not to discuss what happens after CIN, but to explain what causes CIN. Therefore, the 17 CIN features need to be linked to the putative causes of CNA. (Thinking questions, how to use the known to explain the unknown and make people feel credible is the biggest difficulty in Shengxin).
The overall idea of the association:
- First, through the relationship between signatures and CIN-related gene mutations, each signature is given a hypothetical corresponding cause
- Then cross-validate with other datasets of different dimensions
- Then give a score according to the verification result.
usage data
In general, to accomplish this step, as shown in the red box below, they used almost all the public data available today.
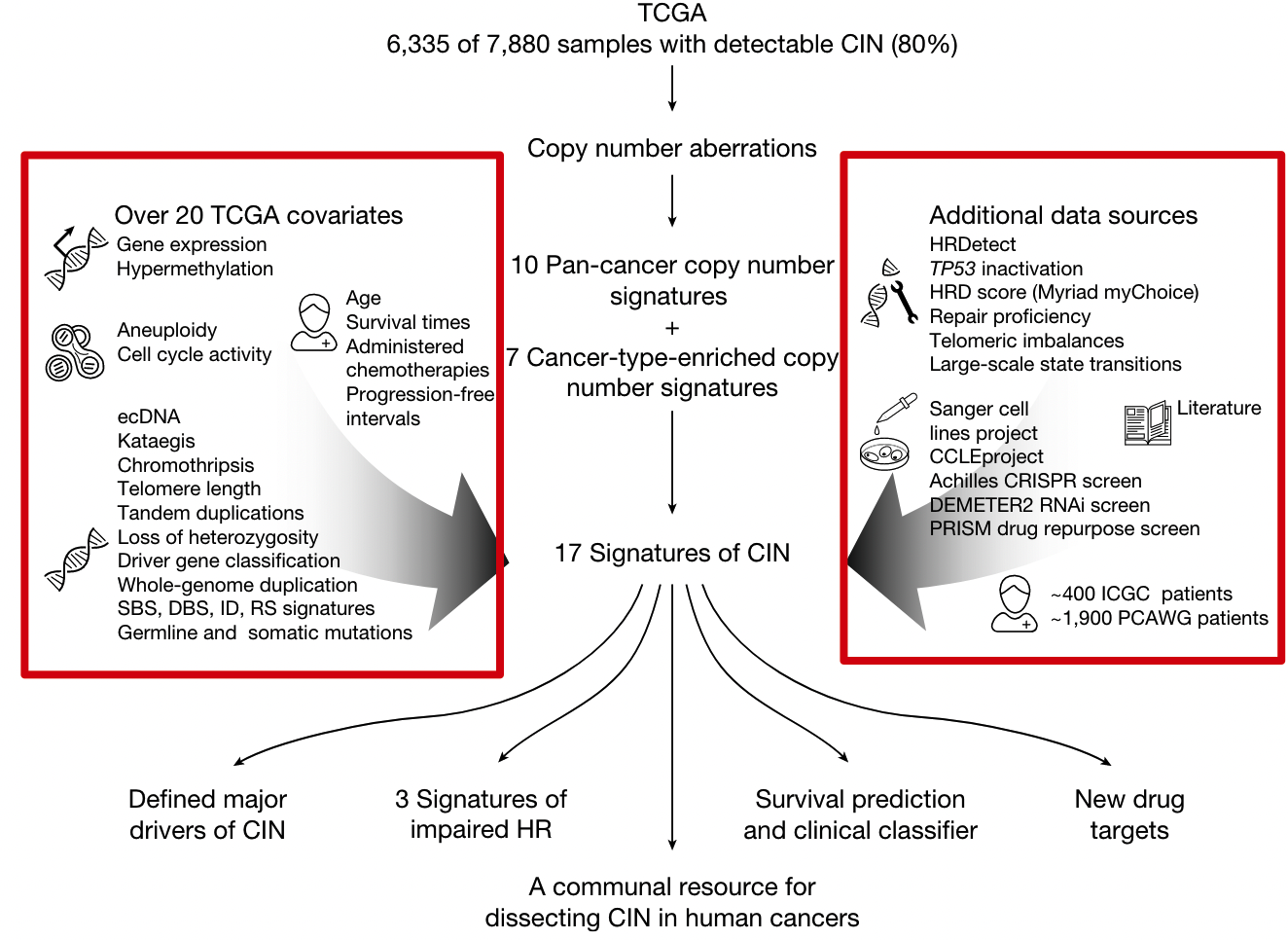
Data from two patient cohorts and their clinical data
- About 1900 patients in the Pan-Cancer Whole Genome Analysis (PCAWG) project
- About 400 patients from the International Cancer Genome Consortium (ICGC) project
Five Mutational Traits
- single-base substitution (SBS)
- insertion-deletion (ID)
- doublet base substitutions
- earlier ovarian cancer research ovarian copy number
rearrangement
14 molecular features
- somatic point mutations
- gene expression
- cell cycle score
- aneuploidy score
- Whole-chromosome copy number aberrations (CNAs)
- tandem duplications
- loss of heterozygosity,
- chromothripsis
- clustered mutant kataegis
- whole-genome duplication status
- telomere length
- elongation machinery activity
- extrachromosomal DNA
- Centrosome amplification score (CA20)
11 DNA repair-specific features
- Germline BRCA1/BRCA2 mutations
- BRCA1/RAD51C hypermethylation data
- HRDetect score
- HRD Score (Myriad myChoice)
- TP53 inactivation score
- telomere imbalances score
- large-scale state transition score
- loss of heterozygosity score
- DNA repair proficiency score
- Protein expression scores for 23 DNA damage repair genes
- PCAWG structural variants with homology
Identification of related CIN genes
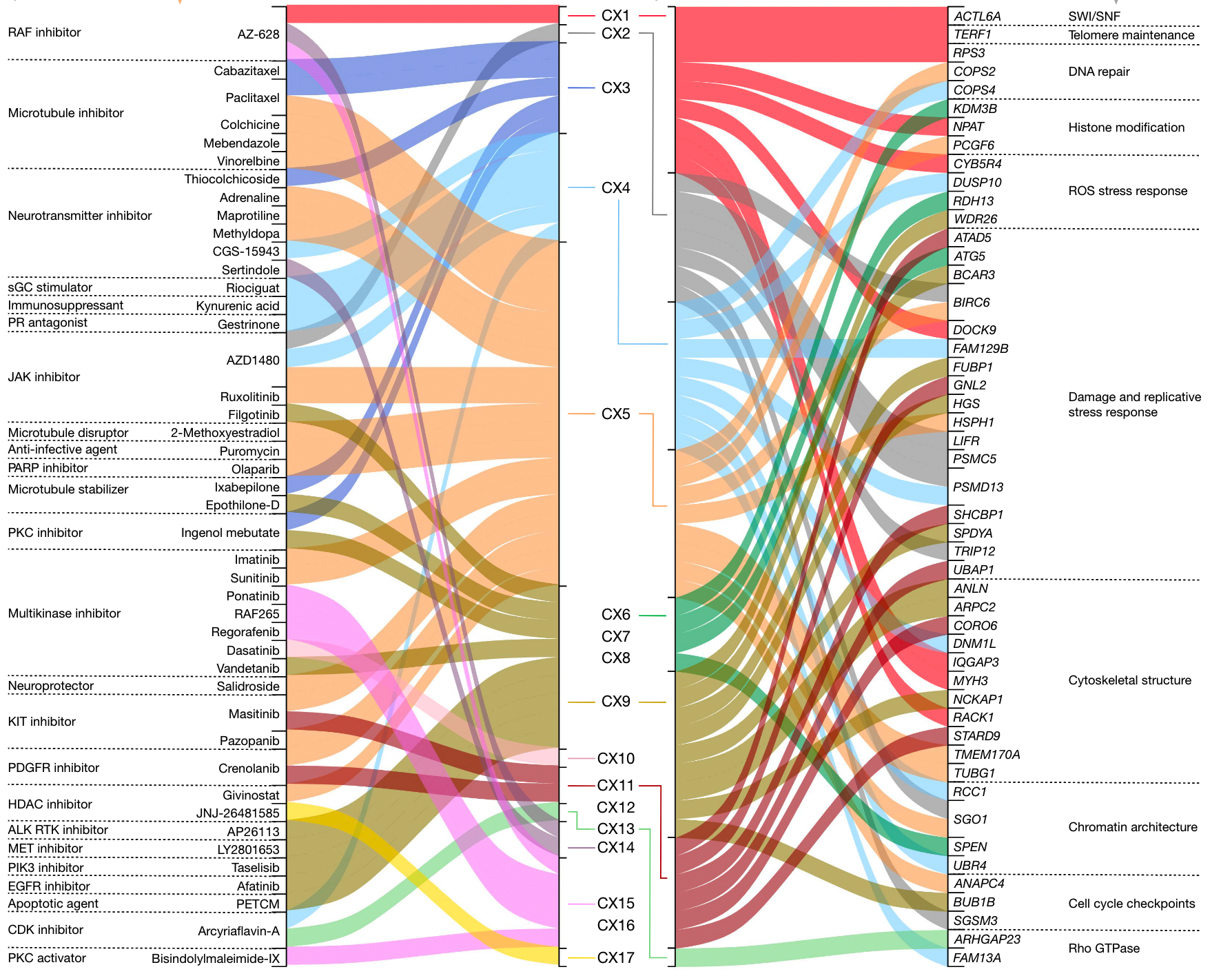
To correlate signature activity with mutant genes, the authors performed the following screens. First, 200 tumor-related genes were selected through previous research on driver genes, and 1330 CIN-related pathway proteins were screened according to the reactoma database. The screened 1179 genes were divided into two groups according to the presence or absence of mutation to detect the correlation of 17 CIN signatures, and found 788 genes related to at least one signature. The 788 genes were filtered by qvalue and effect size, and 32 high-confidence genes that were highly correlated with signature were retained.
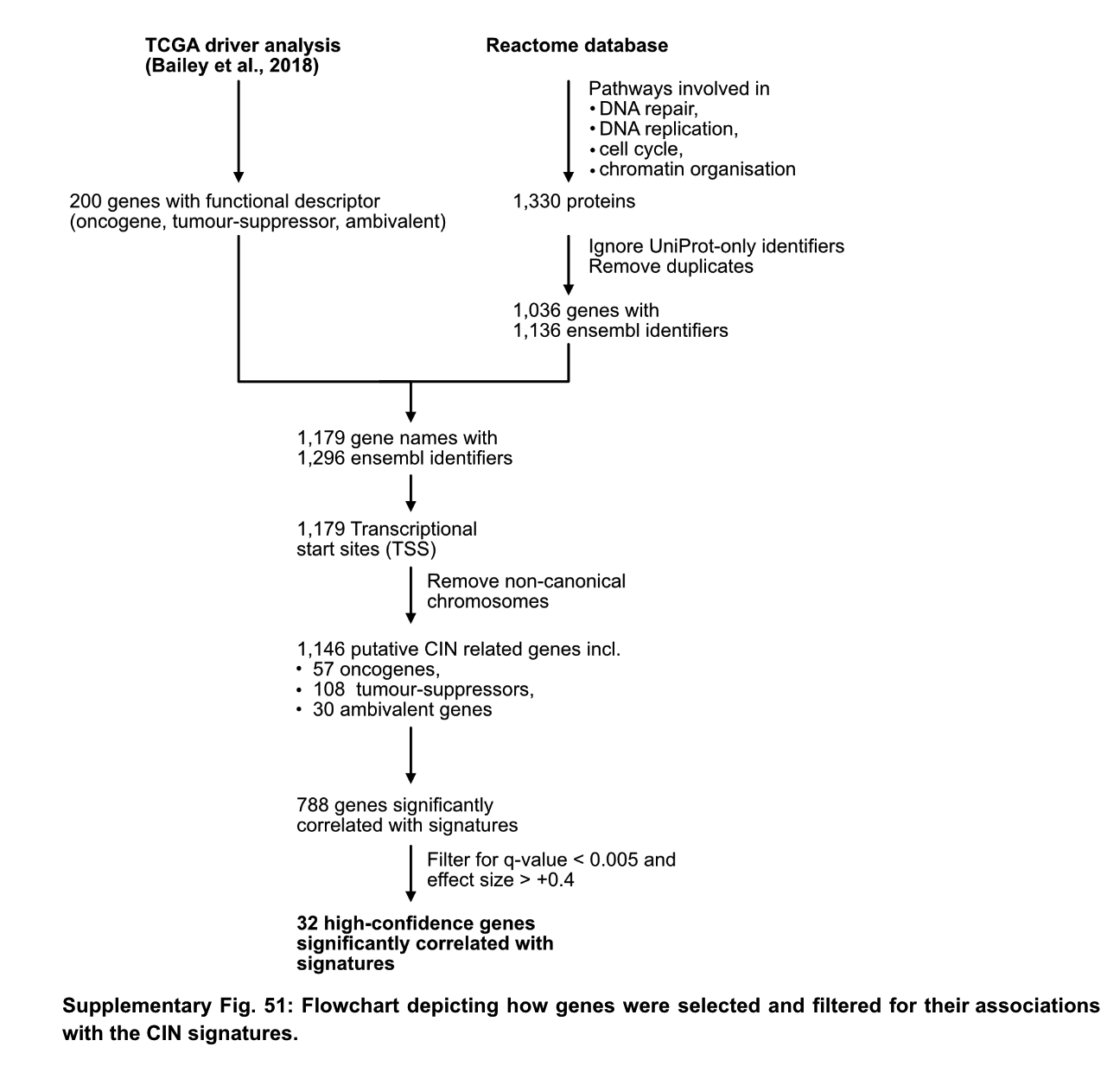
The 32 genes are AKT1, BRCA1, BRCA2, CCND1, CDK4, CDKN2C, CIC, CUL1, ERBB2, ERBB3, ERCC2, FBXW7, H3F3A, IDH1, IDH2, MAPK1, MYC, NFE2L2, RAC1, RPL22, PBRM1, PCBP1, PIK3R2 , PMS1, PPP2R1A, PSMA4, SPOP, SOS1, SOX9, TP53, U2AF1, VHL. The authors examined the literature surrounding these genes and compiled information on how they affect genome stability.
There is a relationship between these gene mutations and copy number signatures as follows
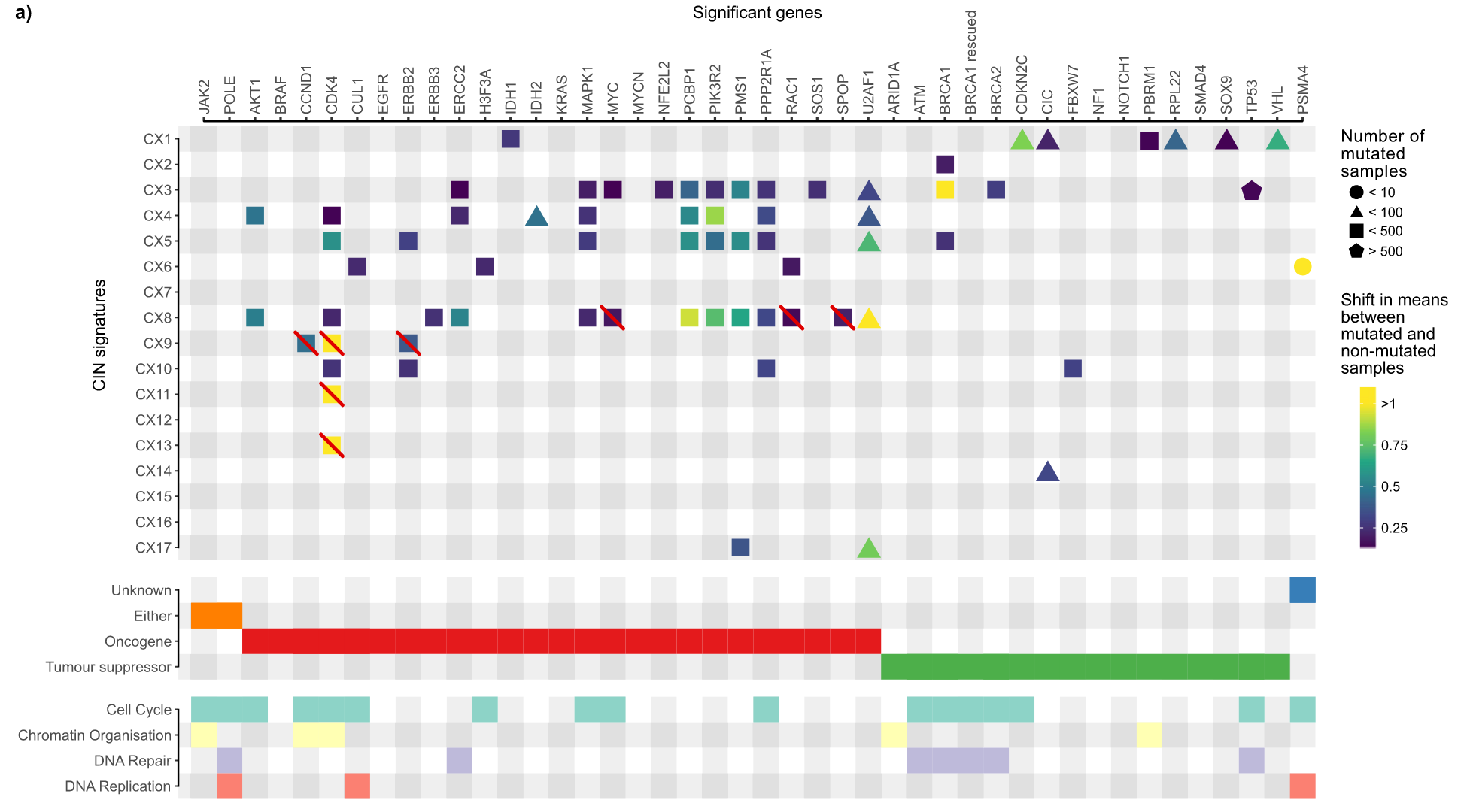
In the picture above you can see that some genes are crossed out by red lines. Why is this?
For the four features (CX8, CX9, CX11, CX13) that show amplification patterns as shown above, the authors investigated their relationship to extrachromosomal DNA amplicons. Since oncogenes are easily amplified by ecDNA, the authors hypothesized that genes amplified by ecDNA may be the result of the processes that generate these amplicons and thus may not be causally related to the mutational process. The changes in the signature activity of the high-confidence genes amplified in the ecDNA amplicons were tested with the data of the relevant research articles, and each high-confidence gene in the amplification-related signature was tested for the amplicon with the gene amplicon. Whether the sample has significant signature activity compared to the sample without amplicon. Several additional genes for hypothesized CIN genesis were therefore deleted.
Etiological hypothesis
The etiological hypothesis logic associated with signature and CIN is shown in the figure below.

If a signature has no genes associated with it, there is no way to make assumptions. If a signature does not include amplification but has an associated gene, look for the mechanism related to this gene and CIN, and give a hypothesis if there is a known mechanism; if a signature includes amplification and has a gene association, first determine the gene Whether enriched in ecDNA as well, if enriched no association is assumed. Then there are the remaining genes, which are hypothesized according to the related functions of CIN. A total of 11 possible putative causes of signatures were obtained using this analysis.
With these possible reasons, use as much external data as possible to verify and give scores.

Add two stars if a gene with high confidence associates a trait with a known CIN type. We add a third star if there is additional data to support this putative cause.
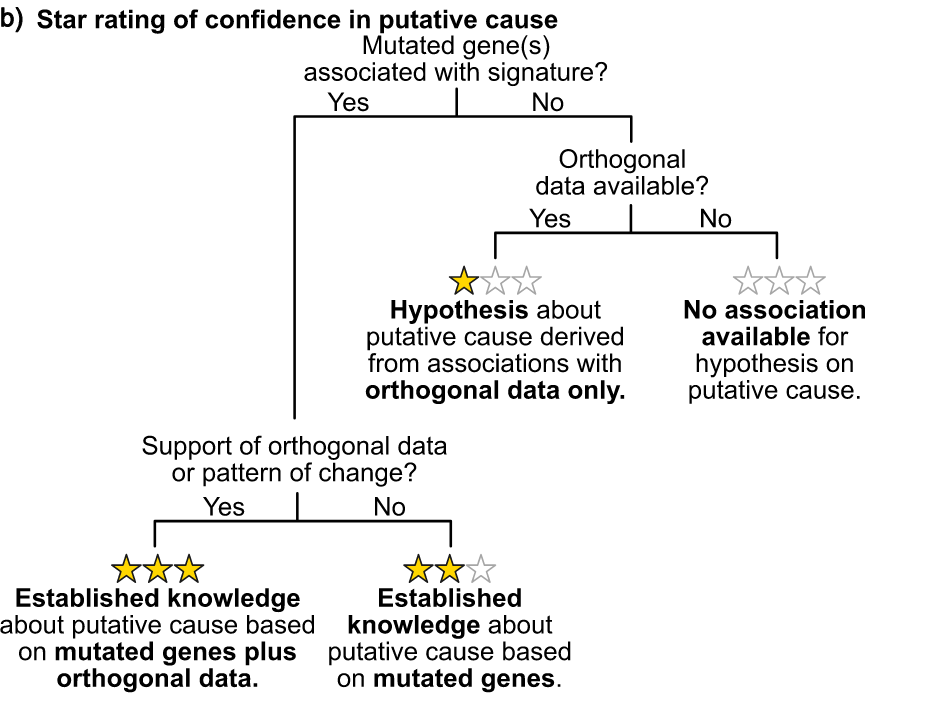
In the end, eight signatures (CX1-CX6, CX8, CX11) received three stars. Three signatures (CX10, CX14, CX17), that is, at least one high-confidence gene associated with known CIN types, but no additional data support. Three signatures (CX9, CX13, CX16) were awarded one star for which no high-confidence genes were found, but some additional data validation allowed the authors to propose a putative etiology. Three features (CX7, CX12, CX15) are sporadic.
A specific example can be seen here. For example, in CX3, the mutational characteristics of itself put forward the hypothesis that it has LST and may be related to HRD; at the same time, a copy number change shows that it may be LOH events and tandem duplications.

Based on this, according to the associated genes, it is found that the functions of the following genes are indeed related to the function of CIN, and first give two stars. So is there any other data to support it? It can be seen that various other HRD-related signatures exist, and are supported by survival data, and finally three stars.
After such a complicated verification, we finally got a core result like the following figure. For a detailed explanation of the causes, please refer to the original text.
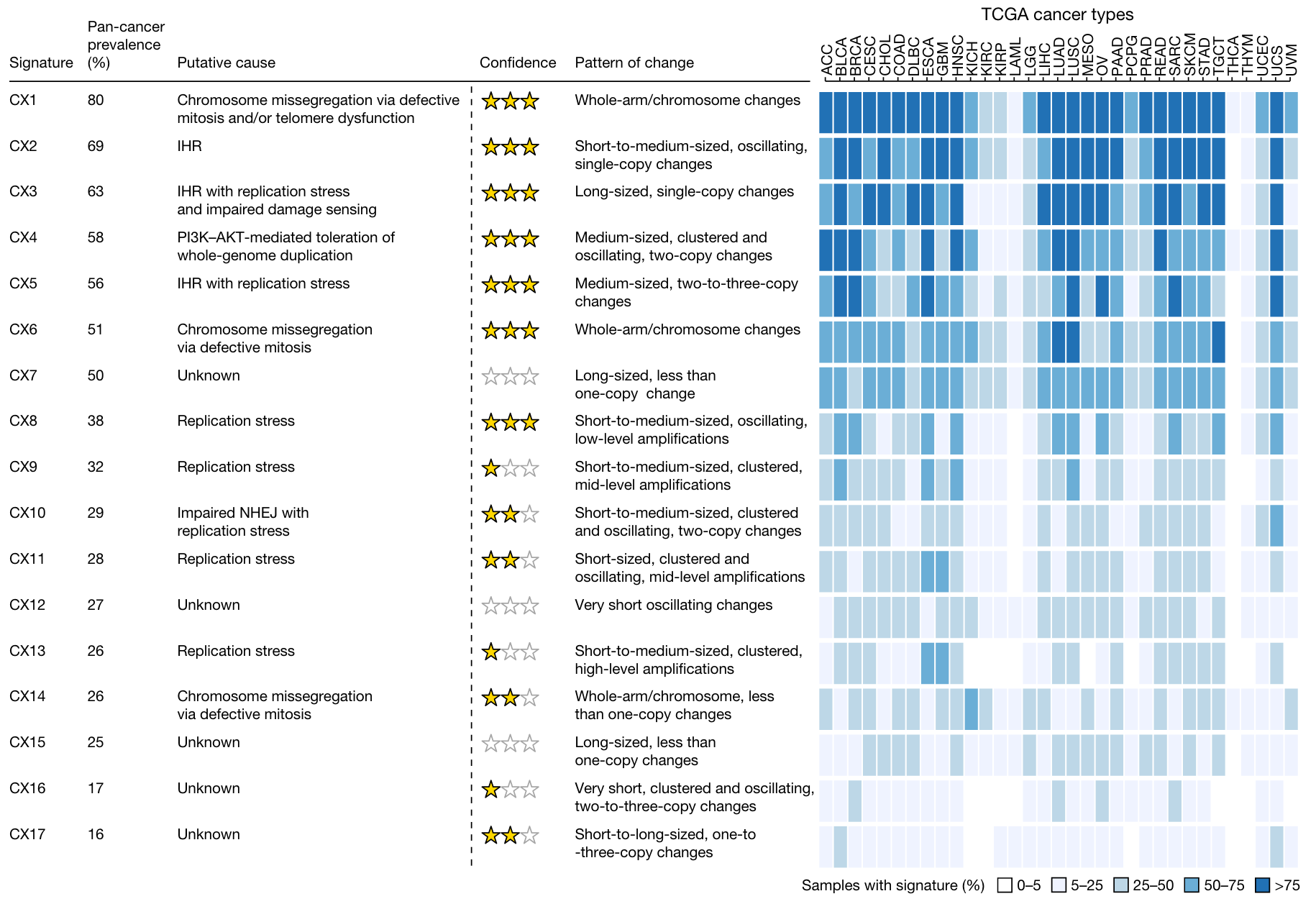
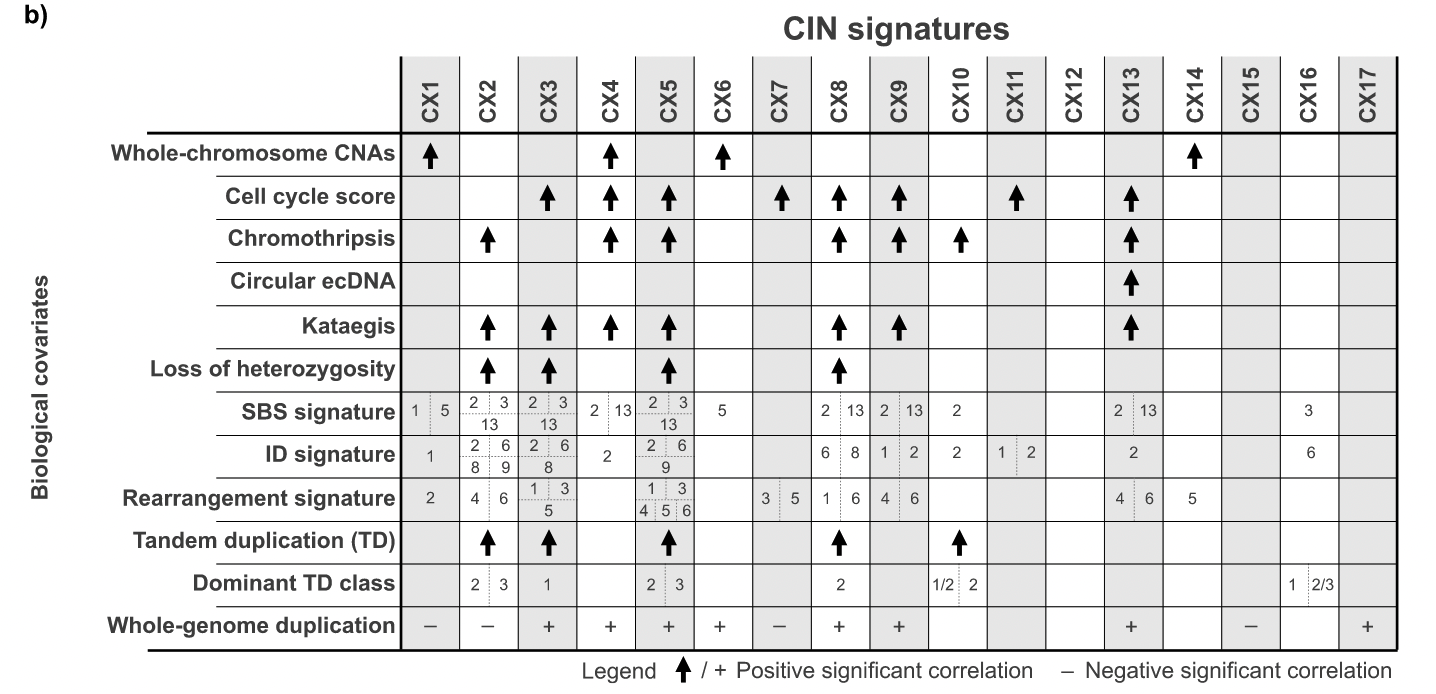
Chromosomal abnormalities were associated with seven distinct features, suggesting that many potential causes underlie these complex rearrangements. Replication stress is associated with eight signatures, highlighting that it is a major source of CIN. Different signatures show a bias that occurs before WGD (CX1, CX2, CX7, and CX15) or after WGD (CX3, CX5, CX6, CX8, CX9, CX13, and CX17), showing the importance of WGD events in regulating CIN . APOBEC mutations and kataegis features were associated with six signatures, emphasizing that these are a common feature of CIN.
Drug Response and Drug Target Identification
With an explanation of why, what can these signatures be used for?
The aforementioned causal exploration results suggest that typical cancer-related sympathies are some of the major drivers of CIN. And many genes in these pathways are the focus of targeted therapy.
Therefore, given that these signatures can be readily measured in patient tumors, the authors next explored their utility for treatment response prediction and drug target identification.
The authors integrate data from 297 cancer cell lines, correlate gene importance determined by CRISPR-Cas9 or RNAi screening with responses to drug perturbations, and discover corresponding biomarkers and novel drug targets. Correlations between signature activity and gene and drug susceptibility were assessed.
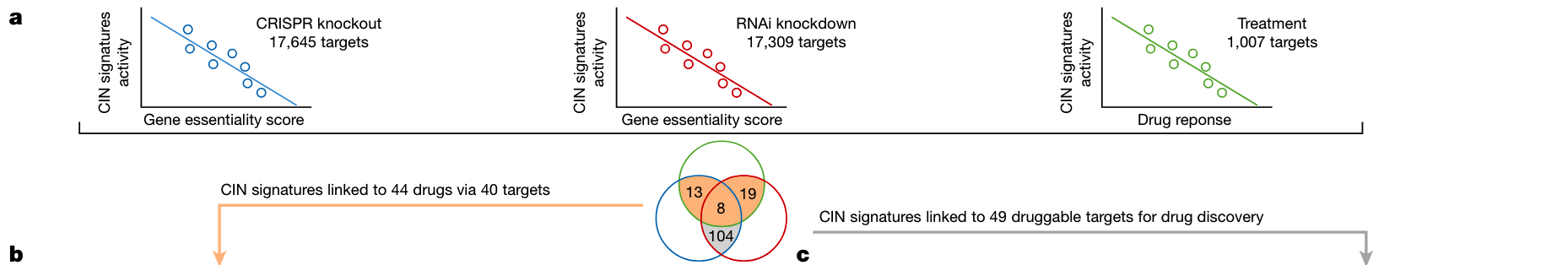
The analysis results are shown in the figure below. The authors determined that the copy number signatures of 40 genes were significantly correlated with the genetic and drug perturbations of the targets.
Among these genes, CX5 is a HR-related signature predicted to respond to olaparib through inhibition of PARP1, and because this signature is also associated with RNAi knockdown of PARP1, it may represent a biomarker for conventional Inhibition of protein function rather than capture of PARP.
CX9 (replication stress-related) is associated with response to multiple kinase inhibitors against genes involved in major mitotic pathways (EGFR, JAK1, MET, PRKCA, and PIK3CA), suggesting that a multi-kinase inhibitor approach may be suitable for replication stress-related tumors .
At the same time, from the CRISPR and RNAi perturbation screening, 104 target genes with pharmacologically acceptable structures were found, and these genes currently have no targeted therapy in the clinic. These represent predicted synthetic lethal drug targets, 49 of which have evidence of involvement in CIN-related mechanisms (shown above).
Predicting platinum sensitivity
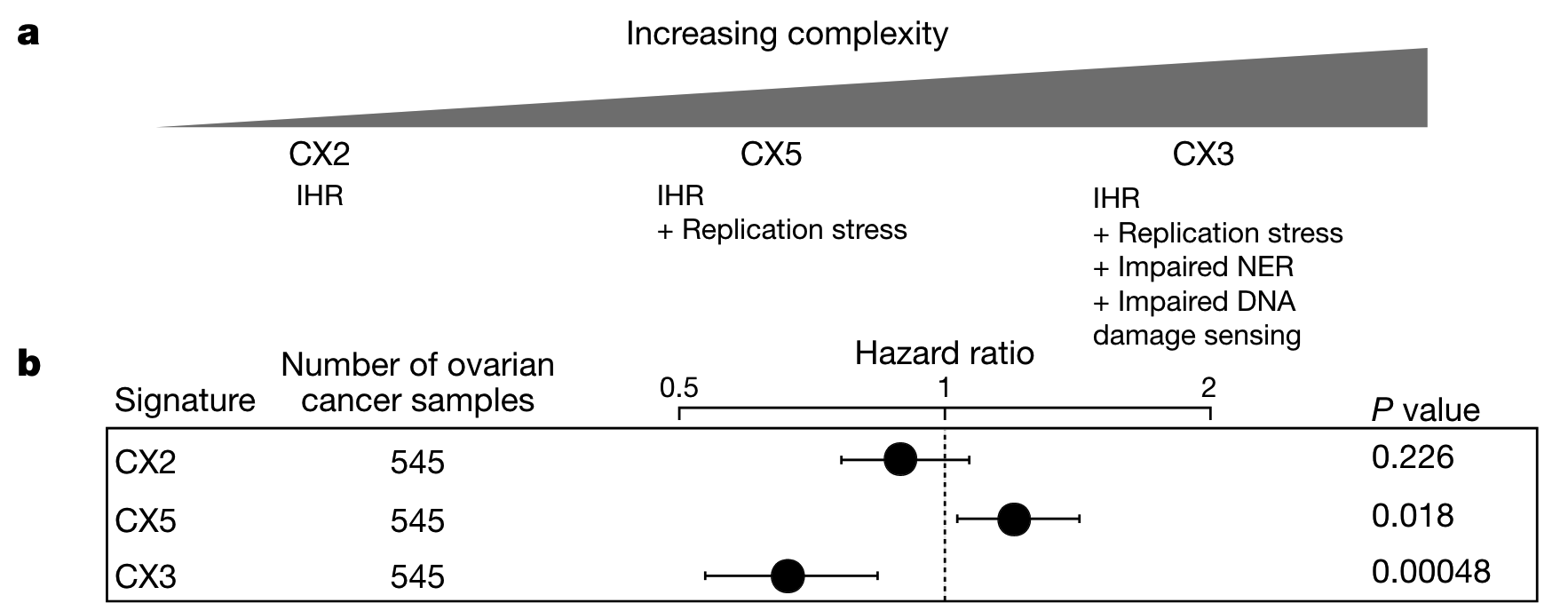
Three impaired homologous recombination (IHR)-related signatures indicate a pattern of increased CIN complexity.
That is, IHR alone produces CX2, which represents a small copy number change characteristic of tandem replication. IHR plus replication stress leads to CX5, which involves larger CNAs. Finally, IHR coupled with replication stress, impaired NER, and impaired DNA damage signaling produce CX3, whose largest CNAs are closely associated with loss of heterozygosity. However, the findings do not indicate whether these different levels of complexity develop in a progressive fashion or as separate processes.
Impairment of HR and NER is known to be sensitive to platinum chemotherapy, and given that only CX3 is associated with disruption of NER, the authors hypothesized that IHR signatures may exhibit different predictive powers of platinum sensitivity.
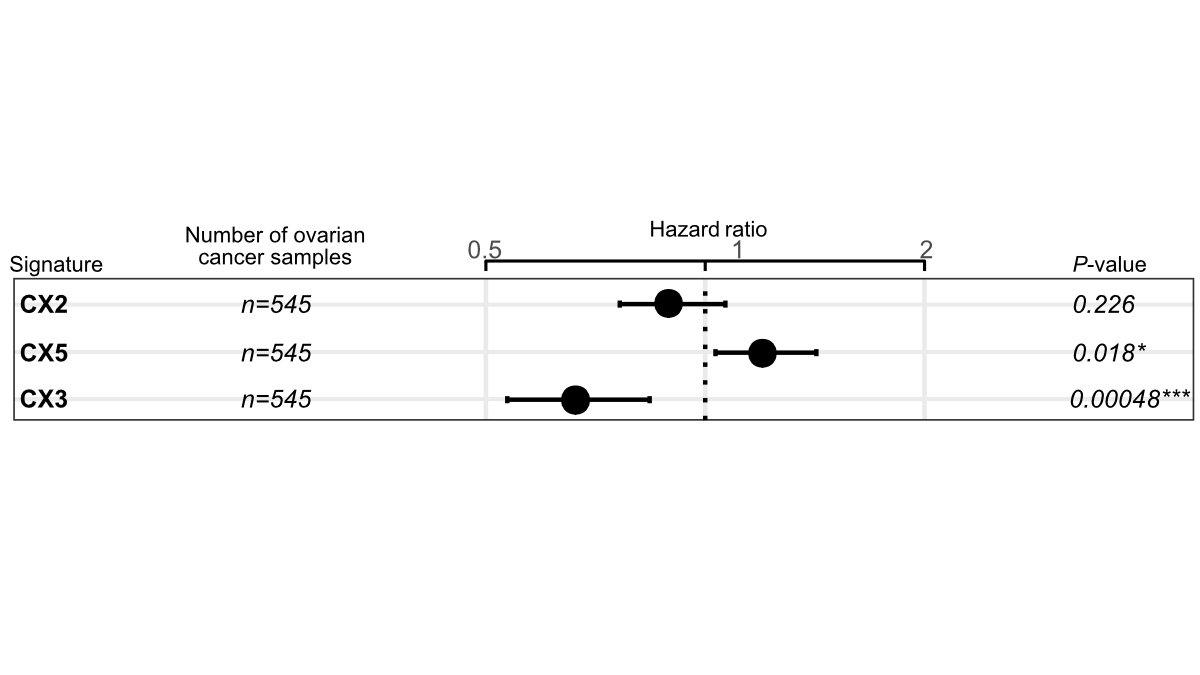
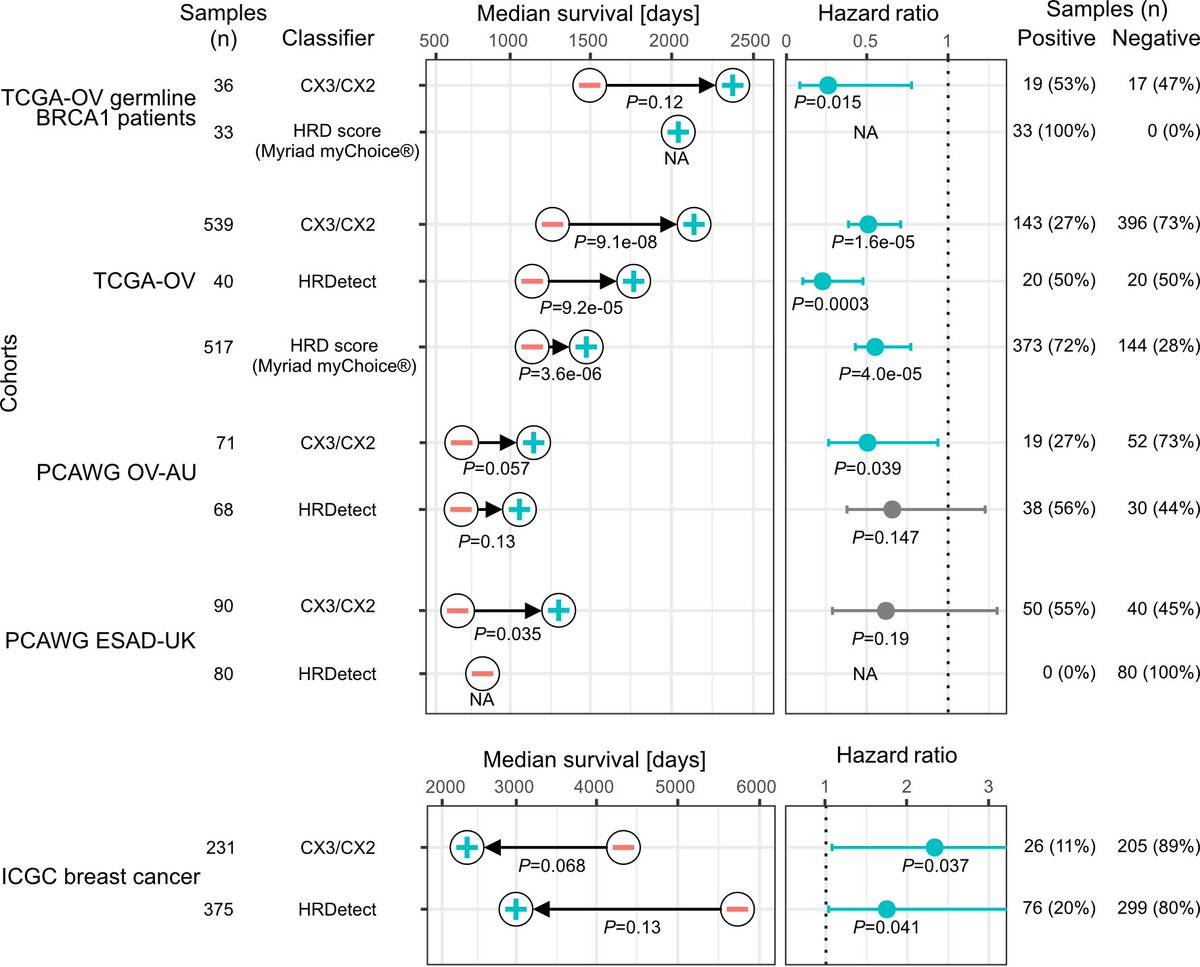
Since ovarian cancer patients often receive platinum-based chemotherapy, Cox models were used to test the ability of three features to predict overall survival to infer platinum sensitivity. It can be seen that CX2 has no relationship with platinum sensitivity, CX5 can predict resistance, and CX3 can predict sensitivity.
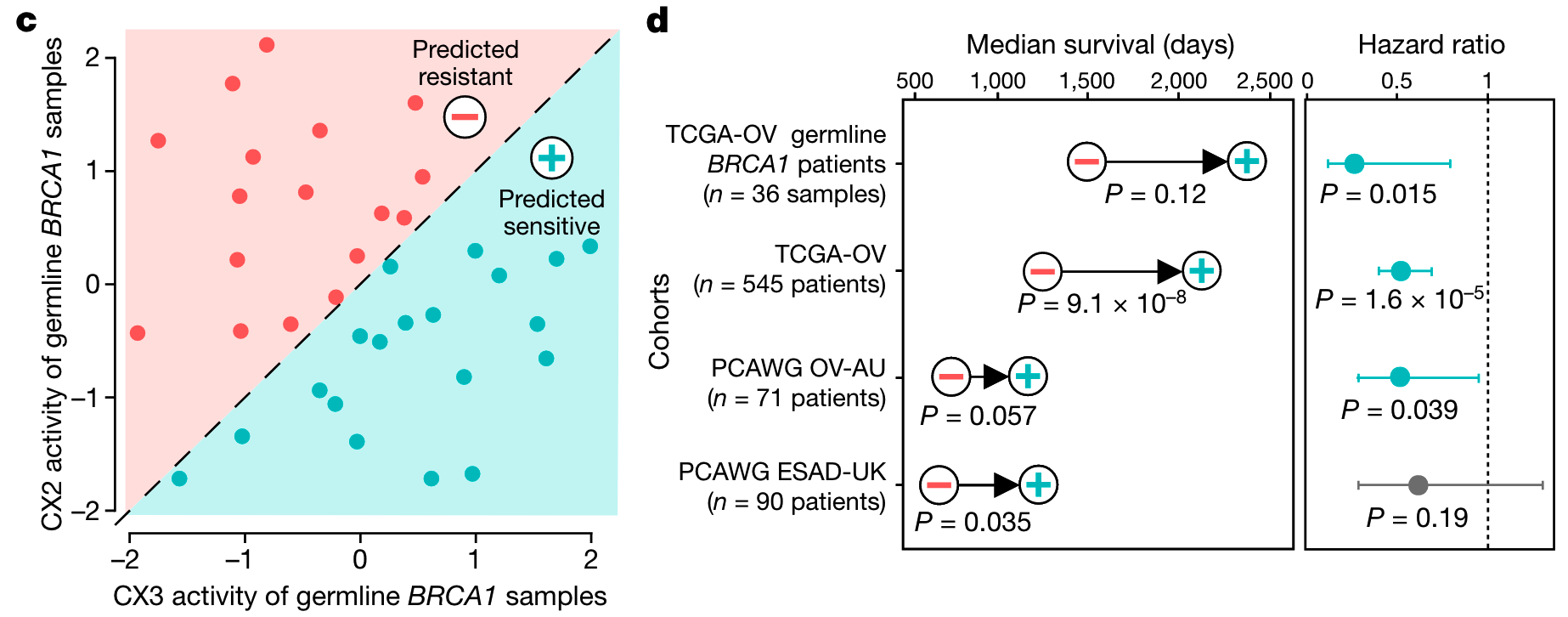
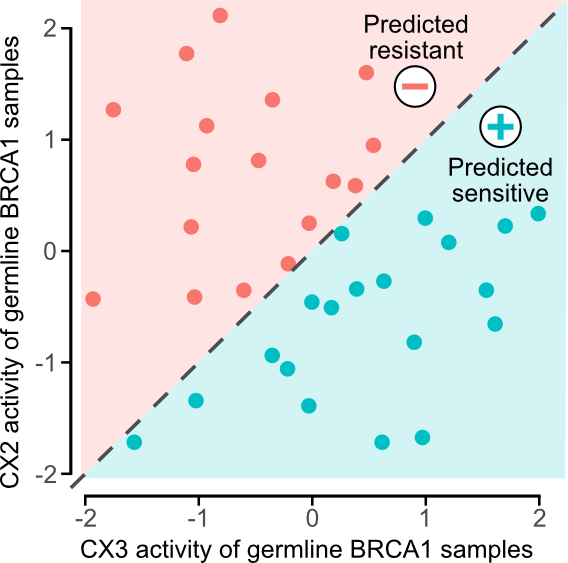
Given that these IHR features are predictive of platinum response, the authors further hypothesized that combining these features could provide better predictors of platinum sensitivity?
Since CX2 is not predictive, the authors first used it as a reference to capture non-predictive IHR-related genomic changes and required predictive CX3 activity above it to confer sensitivity, i.e. if CX3 activity was greater than CX2, then predicted sensitivity .
As shown in the figure below, this interpretable classifier was able to distinguish overall survival in the BRCA1 germline-mutated ovarian cancer cohort, the ovarian cancer cohort in the TCGA cohort, and the independent validation cohort and the esophageal cancer cohort also routinely using platinum-based chemotherapy. In fact, there have been many complex models based on machine learning for prediction before, but the author’s test found that this simple classification does not need to be poor before requiring more information.
The above is the main content of this article, because there are too many more than 60 pictures and 60 pages of attachments, and my combing ability is also limited. If you have other questions after reading this, you have to read the original text yourself.
Next, let’s talk about something that might interest you.
Repeatedly breaking the head with the reviewer
As mentioned at the beginning of the article, it took nearly two years for this paper to go online from submission to online. For a paper without wet test, the pull from review is not too long.
After four rounds of overhaul, I felt my blood pressure rise several times just by reading their 92-page record of the confrontation. As the title says, in 2022, using TCGA data to study chromosomal instability can still publish Nature, why? This is not my question, but the question of several reviewers.
If you really want to improve your scientific thinking, I think maybe reading Peer Review File is more valuable than reading the original text.
first round of comments
First of all, it is worth congratulating that this article was not directly rejected by the editor when it came up, but luckily it reached the hands of the three reviewers.
By reading the Peer Review File, you can see that after the first submission of the review, among the three people, the first one raised 8 major points (Specific points) and 5 minor problems (minor); the second raised 6 points of overhaul and 4 Minor repairs; the third one directly gives 5 major issues.
Fortunately, this time, the editor should be merciful again, giving the author a chance to overhaul. Here are some of the ones that I find more interesting.

Reviewer 1
His first comment was very rude, to put it bluntly, it meant rejecting the manuscript. He mentioned:
- The fact that 80% of tumors have CIN may be overestimated
- How a particular signature is linked to a given etiology is unclear, and the text is largely speculative.
- It’s not clear what impact such a landmark database might have on cancer prevention or treatment, or if it would be of any use.
- What the author writes is unlikely to be understood by non-specialized readers, so it is best suited for a more specialized journal (do you often see this in your own review comments).
Among the 8 Specific points, the reviewers mainly questioned the following issues.
- What is the difference between this article and the author’s publication in Nature Genetics in 2018, please explain to me
- Do some of the conclusions you found hold in more cohorts you give me a test test
- The consistency between your signature and the previous published results in 2018 is weak. Is it because the data this time is TCGA snp6 array, and the shallow WGS was used before.
Reviewer 2
The comments are as follows. From the response of the second reviewer, it can be seen that the authors of the first edition seem to have only explained the two most common signatures in all cancer types, and these corresponding biological processes have been studied more. It is believed that more attention should be paid to other signature interpretations.

The main doubts he mentioned include
- Hopefully the authors can compare CX2 with other copy number based HRD signatures such as HRD score and HRDetect.
- Recent WGS studies have described somatic copy number alterations in great detail at higher resolution than SNP arrays (in fact, PCAWG). Can the authors compare with recently published results?
- Since the biological interpretation of CIN features is paramount, how can these CSs be identified based on WGS? The ability to detect these is tricky because the number of fragments and their size can vary.
- The method of signature derivation is not clearly written, maybe it can be illustrated with illustrations or flow charts.
Reviewer 3
When he broke his head for the first time, it seemed that the evaluation was very restrained, but in fact, he was the most suspicious, which also caused the introduction of a fourth reviewer in the following rounds.

The third reviewer felt that while the idea that CNV signatures could inform understanding of CINs is promising, the methods described by the authors are not novel or informative enough, and the findings lack solid supporting evidence.
His main questions include the following:
- Many of your conclusions are based on correlation coefficients. You surely know without a doubt that even a weak relationship can become statistically significant when there is a lot of data. For example, you yourself said that there is a correlation between 5-year survival and CIN, but it is clear from Figure 1b that the correlation is driven by a small number of outliers, I am surprised that you claim CIN based on this figure associated with survival. (It seems that the author of the first edition of the article put a correlation between survival and CIN at the beginning, and was directly dissed by the reviewer. It can be said that it does not hurt much but is extremely insulting)
- The study does not describe how this paper differs from or complements other papers on the same topic, especially considering that the SNP data are very old and have been analyzed before (simply put, what’s the point of your matter).
- For the IHR signature, the length of the copy number segment (4.9-30.8Mb) appears to be much larger than that reported for the SV signature (1-10kb) (Nik Zainal, Nature 2016), it is really unexpected that HR defects are as common as reported in this study outside. You yourself state that nine different signatures are associated with homologous recombination defects without providing any mechanistic explanation. I don’t believe it anyway.
- Totally don’t understand why you don’t extend your analysis to WGS and demonstrate that the findings are reproducible and consistent with the existing literature.
- To determine whether a CIN is present in a sample, the authors used a somewhat arbitrary threshold of 20 CNA, which may have exaggerated the proportion of samples with actual genomic instability. Again, the same samples (not only SNP arrays, but also WGS) have been analyzed before, but no comparisons or checks have been made on the robustness of the conclusions for these parameters.
- Should take another look at possible overfitting and false positive issues. You are testing the necessity of applying a minimum threshold to both signatures. However, these two are associated with whole chromosome or chromosome arm copy number changes and are more easily detected. Will there be false positives for others?
Let’s summarize here, the most important thing for a few people is the two ultimate tortures:
- Is your thing applicable to multiple sequencing methods?
- What is the clinical use of yours?
What did the author update
Everyone has experience when submitting manuscripts. Questions about reviewers need to be answered one by one. If you are interested, you can read them yourself. Only a few of the most critical revisions are mentioned here.
The author’s words are: This revision includes significant improvements and additional results, and we have completely changed the content of the article. A translation is a rewrite.
- The robustness of our method was validated by comparing samples from the same patients across five sequencing platforms. We also define a noise threshold for specific signatures, which makes the detection of low activity signatures more robust and avoids overfitting. (In fact, readers will also raise this question. I wonder if you have this doubt when you read the previous article ?)
- Causes were determined by combining associations with mutational driver genes, copy number patterns, and multiple external datasets. Multiple sources of evidence for each hypothetical cause, including star ratings, are more clearly stated. We present all evidence in web resources for readers to explore. (Completely rewrites the content related to causation speculation)
- Highlight the clinical impact of our approach: (a) use our signature to identify drug targets and predict drug response; (b) develop a signature-based classifier to predict platinum sensitivity in ovarian cancer. (It turns out that these two parts did not exist when the first submission was made)
- Substantially expand our integrated data and resources to include PCAWG and ICGC cohorts, additional independent annotations and variables, and measures of different types of CIN.
Although we can’t see the first edition of the article, it can be seen from these revisions that the author dares to directly submit to Nature for the first time. The big deal is to be a person who has been rejected by Nature.
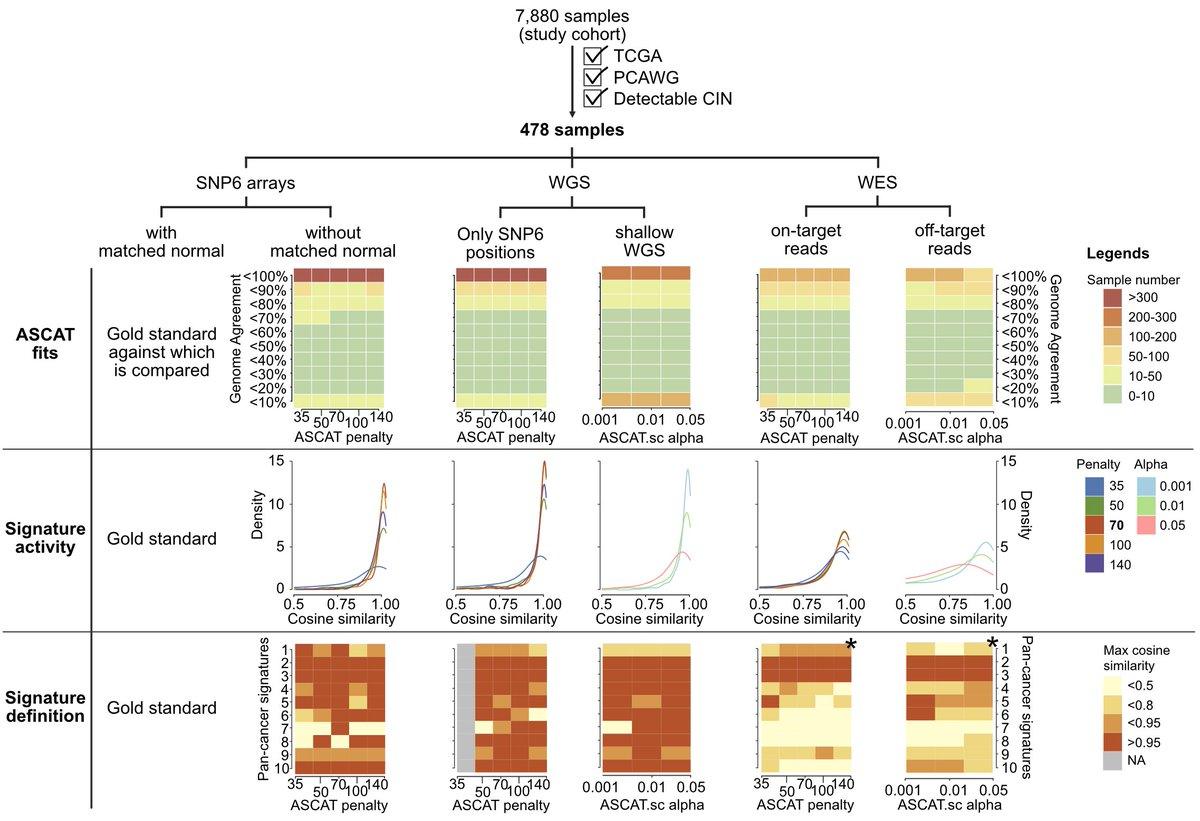
Regarding the robustness test, in fact, in the final article, the author has a lot of analysis in the attachments and drawings to confirm. In order to verify the robustness of this signature model, the authors screened out 478 samples with PCAWG WGS data in TCGA, performed downsampling based on SNP6 chip and shallow WGS downsampling for WGS data, and differentiated WES data into on-targets and off-target, for consistency evaluation.
Five chromosomal instability activities were also simulated by introducing copy number changes in the human genome:
- Chromosomal misplacement (CHR) via mitotic errors
- Large-scale state transition (LST) induced by homologous recombination defects
- Large fragment amplification (ecDNA) by ecDNA circularization and amplification
- Early genome duplication through failure of cell division (WGD early)
- Late Whole Genome Duplication (WGD late) via endonuclear duplication
Then use your own method to verify whether these contents can be restored.
The first edition of this part of clinical application value seems to introduce a correlation between CIN and survival in fig1. This time the author’s reply is to admit it, so we will delete this part of the content. Then if you want clinical value, then I will give you a good clinical value, so that you can see the drug prediction and chemotherapy sensitivity prediction above.
second opinion
After the author replied, the second wave of attacks ushered in.
Reviewer 1
The first was slightly persuaded and re-raised 6 small questions

Reviewer 2
The second person also affirmed, but said that because you have rewritten an article, then I will add 6 small questions based on your newly written article.

Reviewer 3
After the attitude of the first two reviewers changed, the third reviewer began to attack.

You can get a feel for what he roughly said, the following is a rough translation.
The authors have done extensive work since the first submission, including analyses of several additional human cancer cohorts and cell line models. They developed a CIN signature-based classifier and a model to predict platinum chemotherapy in BRCA1-mutated ovarian cancer. This is an interesting observation, but due to the small number of cases in WGS, it is impossible to conclude whether HRDetect can achieve this goal (could he be the author of the HRDetect method?).
The authors also compared different sequencing platforms to assess the robustness of their CIN characterization method, but these comparisons are based on downsampled WGS/WES data, and it is unclear whether their method still remains when applied to raw WGS/WES data. Has the same degree of robustness (is it dissatisfied that you didn’t pay to test new samples?).
Despite extensive additional work, we do not think the main conclusions of the new version are convincing. We believe that there are serious methodological problems with this article, and we find the causal explanation for the signature to be speculative. Therefore, we consider this article inappropriate for publication in Nature.
As to why there are serious methodological problems and why it is not suitable for publication, he wrote a long content and many questions, three of which are as follows:
- There are flaws in the signature design method, which is not as rigorous as the previous SNV signature method. Specifically, a CNV will contribute multiple features at the same time during calculation.
- Or tell stories based on relevance. In the first version, you were related in a single dimension, and now you become related in multiple dimensions, but still related. For example, the enrichment of CX2 and CX5 in tumors with mutations in the homologous recombination pathway does not imply that these features are specific or due to impaired homologous recombination.
- Many analyses have been done on HR-related content, but these are contradictory, such as CX5 being associated with PARPi sensitivity but also platinum resistance.
What did the author update
In this wave of operations, the author must have gained a lot of confidence, and did not rewrite the article again, “just” added 7 figures and 6 tables.

Here we focus on picking a few answers to the questions related to Reviewer 3:

Regarding the comparison of CIN signature and HRDdetect for the sensitivity of platinum chemotherapy in patients with BRCA1 germline mutation, the author said that HRDdetect only has three available samples, you are not saying that the effect is not visible because of the small number of samples, then I will directly remove it from the text By the way, by the way, the HRDetect algorithm uses the BRCA1/2 mutation as a positive label during training, and we don’t expect it to do anything in the mutation group.
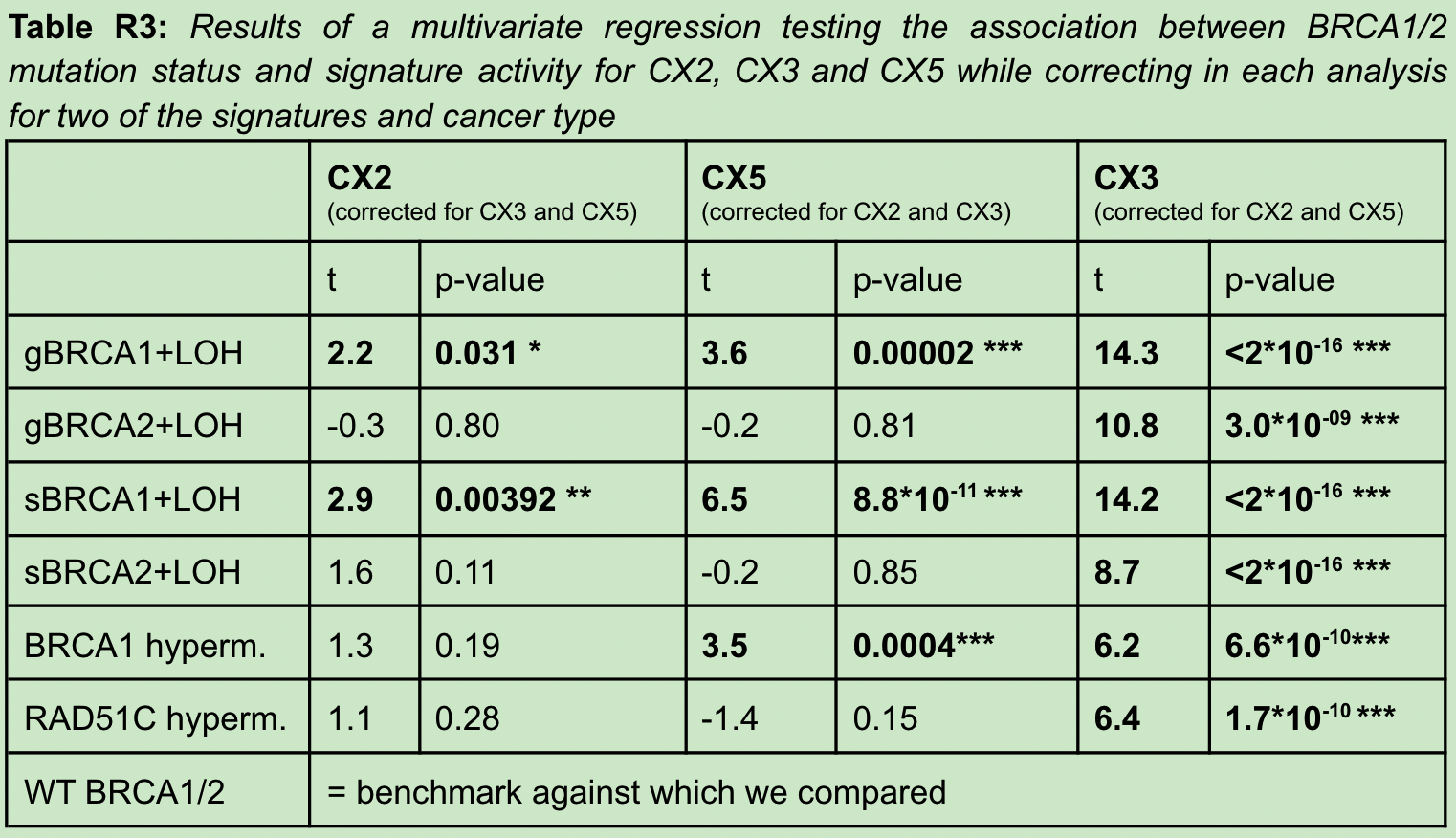
For the reviewer to question CX2 and CX5, it may not mean that these features are specific due to impaired homologous recombination, but a by-product. The author cleverly uses the three signatures of CX2 and CX5, CX3 and cancer in the A multivariate analysis was performed between samples of various BRCA mutation types. demonstrated that after multivariate adjustment, CX2 and CX5 were then significantly different in BRCA1-mutated samples, thus suggesting that CX2 and CX5 may be directly related to BRCA mutational status as a result of impaired HR.
When the reviewer questioned that CX5 is related to PARPi sensitivity but also contradictory to platinum resistance, the author gave the following answer.

It’s an interesting observation (the implication is that you’re looking carefully), and it does seem counterintuitive since it’s accepted that platinum sensitivity also means PARPi sensitivity. CX5 appears to suggest that some platinum-resistant tumors may also be sensitive to PARPi. This has some support in the literature, where platinum-resistant germline BRCA-mutant ovarian cancers have shown sensitivity to PARPi. However, we felt that we did not have enough data to explore this issue further and did not highlight it in the article (our results are also supported by the literature, you can check the literature more).
third opinion
After the author’s wave of replies, the first two reviewers have already indicated that they can.

Interestingly, Reviewer 2 even replied to some of Reviewer 3’s views, and gave the author an idea by the way, probably because he couldn’t stand it anymore.

Reviewer 3
The pressure came to the third reviewer, and his comment this time can be seen to be “anger”.

- The authors appear to be unaware of previous literature in the field and not fully aware of the mathematical framework they employ.
- Every time I review, I just give an example of the problem in the article, even if you answer my example (such as deleting the original text from the article), it does not mean that the problem is solved.
- The authors said at the outset that they carefully assessed all correlations, but several that we asked for further validation were removed due to lack of support. Most of our problems resulted in the deletion of statements in the paper, which contradicts the claim that the correlation is well supported. (This is really God’s logic. If you have revised the question I raised before, it means that you have a problem)
Interestingly, in this review, this reviewer did two things. He first cited a CNA signature article published in a preprint in 2021, saying that the article did not make the author’s mistake. In fact, the article he cited was another article published back-to-back that we mentioned at the beginning.
Second, in order to prove that the model used by the author is really faulty, he even did a simulation study himself to prove that the proposed method is faulty. A possible solution to this problem is to use another back-to-back article.
At this point, the final pressure was placed on the editor. After three rounds, the three reviewers were two to one, but the third was very tough, so the fourth reviewer had to be used as a referee to enter the final review. Fourth round of revisions.
What did the author update
The author’s reply this time was the most interesting of the four rounds of replies.
In order to let the fourth reviewer understand something, the author first explained what happened in the first three rounds of review. The following is the author’s journey.
At the end of the overall response, the author did not forget to say this:

The topics we discuss with reviewers have become more detailed and technical over time, but it is important not to lose sight of the larger issues. Our study synthesizes large and comprehensive genomic data into a coherent framework to measure chromosomal instability. This framework provides an important step change in deepening the understanding of CIN, a field that has made only sporadic progress over the past few decades! (Hahaha, you are almost done).
The big picture is the big picture. The author answered two main questions in this round, which is the complete end of the discussion with the third reviewer.
Reviewer 3’s first issue was citing an article of the same type that he felt did not make a methodological error.
The author used the second reviewer’s idea of simulating the genome to perform a simulation comparison, setting five different mutation processes, while the other paper gave only 3 correct results.
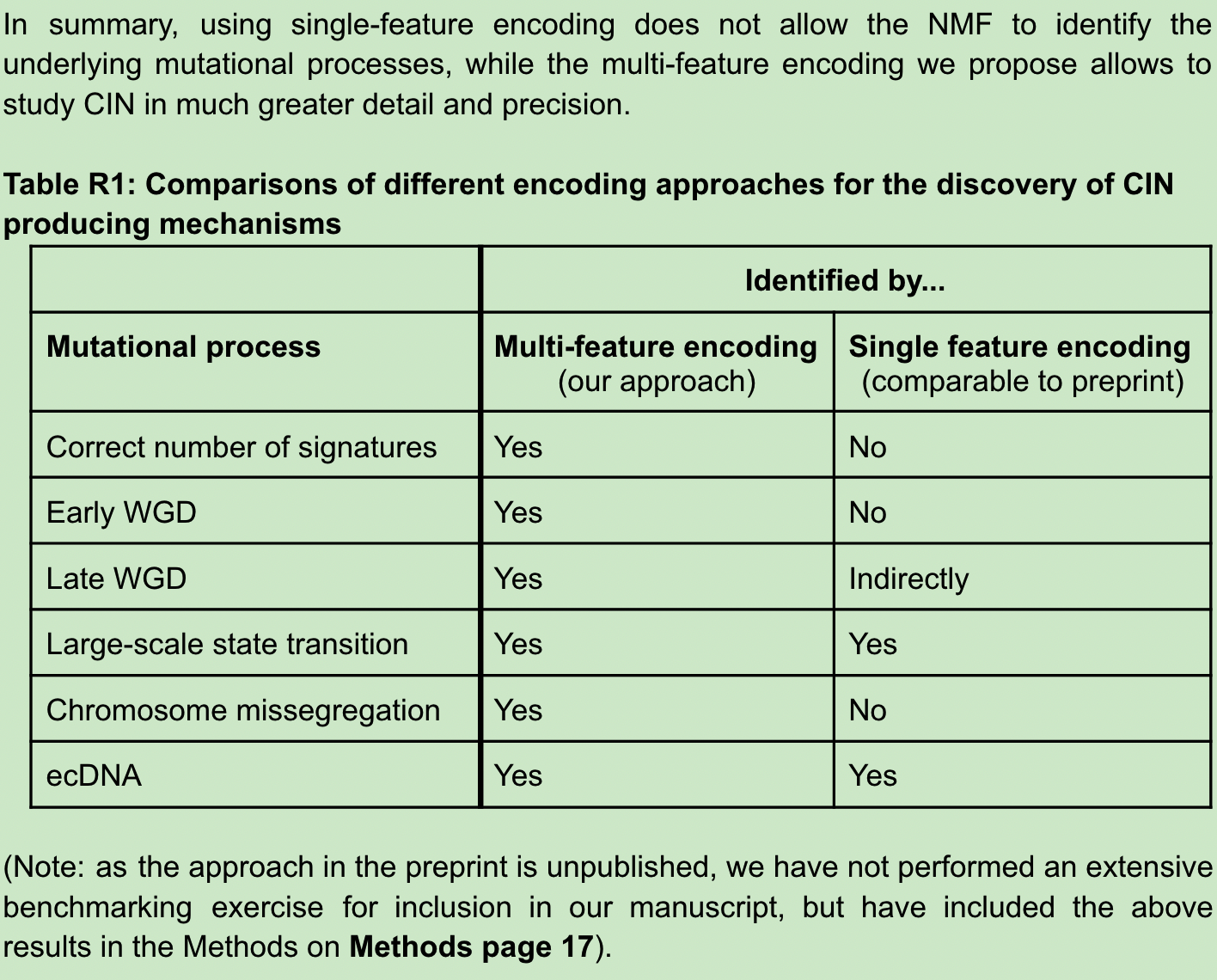
As for the reason, the authors explained that since single-signature encoding uses absolute copy number, the same signature will be artificially divided under different ploidy backgrounds, which is why they deleted the copy number in multi-signature encoding. The three processes CHR, WGD-early and WGD-late are captured by multi-feature encoding because they can represent many biological properties of CNA in the entire feature space. For example, early and late WGD, despite having similar numbers of copy number changes, can be distinguished by the characteristics of fragment size and the number of breakpoints per 10MB. (Writing here, I don’t know what the author of another back-to-back article thinks)
The second problem of reviewer 3 is that he simulated an analysis by himself, and failed to reproduce the input signature using NMF, thus proving that the method has serious problems.
The authors state that the reviewer’s simulation is not an efficient simulation of our method but has a fatal design flaw that artificially causes the NMF process to fail. So another example of a correct simulation is given to verify the results.
At this point, the three rounds of head breaking are over, and the entire review process is basically over.
Fourth round of comments
In the fourth round, only reviewer 4 made some comments, believing that the author has answered the questions of the previous rounds well, and this is an important research that will be a useful supplement to related fields. And he just raised some questions that may need to be revised in the presentation of the article.

Review and revision process
Reviewing the experience of the four review processes, it can be concluded that the author’s article has undergone the following changes.
- The initial submission focused on the derivation of signatures and their biological interpretation, in a way that extended the methodology of a previously published article (Nature Genetics 2018), and a demonstration of the 17 signature-related reasons we found. There is clearly a lack of more data support, robustness proof and most importantly clinically relevant utility here. One reviewer rejected the manuscript, and the other two needed major revisions.
- . The second submission is basically rewritten according to the editor’s request, adding a lot of evidence to the proof of the cause, and adding an important part of the prediction of drug targets and treatment effects. Reviewers 1 and 2 were very positive, but reviewer 3 questioned the basic method (why didn’t you say it earlier?). In response to reviewer 3 looking at our approach from the perspective of previous SNV/SV approaches, the authors point out the unique challenges of copy number analysis.
- Several figures have been added, and after the third submission, Reviewer 3 crystallized their methodological challenge into a simulation study they conducted themselves. At the same time, the approval of the method of another article was proposed, and it was clearly rejected. The author compared and verified the simulation data of the two methods, and once again answered the simulation analysis error of reviewer 3.
- After the fourth and fifth submissions, the author made a lot of revisions to the presentation and details of the article, and finally presented the 68 figures we see now, with 60 pages of attached method descriptions.
Do research and tell stories
By reading the story of the four major overhauls, you may have fully realized the ordeal that bioinformatics methods may go through to publish a Nature in public data, but I have to admit that the probability that you and I should go through this kind of ordeal is very small.
Knowing this, why do I still write 10,000 characters to record this process? In fact, the real intention of reading more than 90 pages of Peer Review File was to try to figure out how a Nature-level research would improve in terms of storytelling and logic from execution to publication.
Luckily, after reading the tortured 90+ pages of the article revision process, I came across the main thread of the article posted by the corresponding author on social media. I finally realized how a fragmented and tinkering scientific research project has become a rigorous and exciting scientific story.
At the end of this push, you should also read it. The following content is from the social platform of the corresponding author. I have roughly translated it, and everyone will focus on the presentation logic of this research at this moment. The content in parentheses is my own supplementary explanation~
a good science story
We have been battling CIN tumors for many years. We frequently see TP53 mutations in CIN, but rarely other drivers. After a while, we began to see patterns in the chaos and began to realize that these characteristics represented different causes of CIN. Can these help us understand CIN better? (submit questions)
Our first success was in ovarian cancer research, where we discovered how DNA copy number (CN) patterns are encoded to allow identification of causal mechanisms. This includes fragment length, number of breakpoints, and more. The authors identified copy number signatures from shallow whole-genome sequencing of high-grade serous ovarian cancer (HGSOC) cases. (Introduction to background)
We have since improved the previous encoding, such as no longer including the absolute copy number of fragments. It seems counterintuitive, but it works surprisingly well. Now, the same CIN types in different ploidy backgrounds are captured by a single pattern rather than many patterns, which enables us to enable pan-cancer analysis. (Introduction to method improvements)
We know from previous studies that each tumor will have multiple types of CIN, resulting in multiple patterns of overlap across the genome. Therefore, we use a Bayesian version of NMF to unravel these patterns with our new encoding.
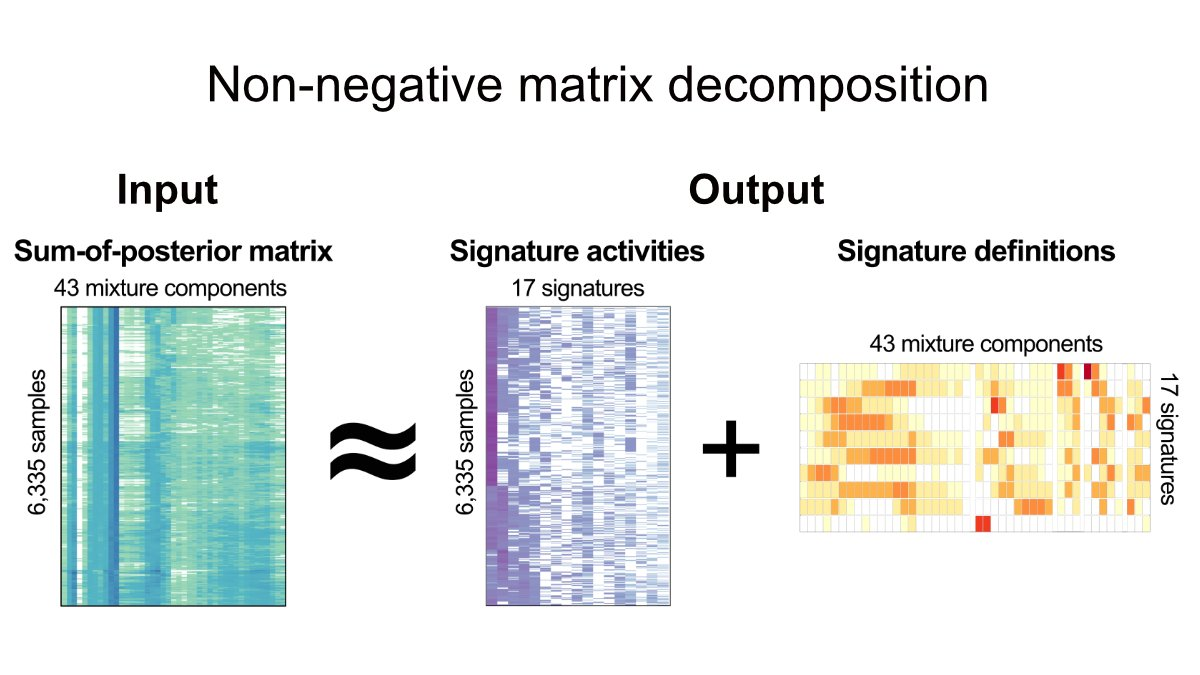
We identified 10 features using NMF in pan-cancer species. This was then applied individually to each large cancer type to identify seven additional pan-cancer signatures. This gives us a total of 17 pan-cancer CIN signatures.
We tested the robustness of encoding and feature identification by correctly identifying combinations of 5 known causes of CIN in mock genomes. These five features include mitotic errors, cytokinesis failure, endoreduplication, homologous recombination deficiency and ecDNA circularisation/amplification.
Our signature is also robust across different genome analysis techniques. Both SNP microarray, deep and shallow whole genome, and exome sequencing. (Content added after the second revision)
Now that we are confident that we have a strong 17 CIN signature, we must climb the next peak: understanding the biology behind these signatures. (Progressive, introduction to the second part explaining biological reasons)
The first challenge: Since NMF produces sparse signature definitions, we need to make them easier to interpret. To do this, we combine the information from the input matrix, activation matrix, and definition matrix to generate an interpretation matrix, where each signature is defined in each decomposition component. (Content added after the third revision)
This matrix allows us to begin to understand our signature, and in many cases, the coding pattern already suggests a mechanism (eg, large length segments + no breaks per chromosome = chromosome misalignment due to mitotic errors).
The second challenge: linking signatures to flawed pathways. To do this, we tested whether patients with mutated driver genes had significantly higher signature activity. These connections, together with the matrix above, allow us to come up with conjectural reasons for the 12 sigantures. (Second revision to improve)
The third challenge. Find supporting evidence for the hypothesized cause. To do this, we integrate a large amount of data to validate and refine our speculation. (Second revision added)
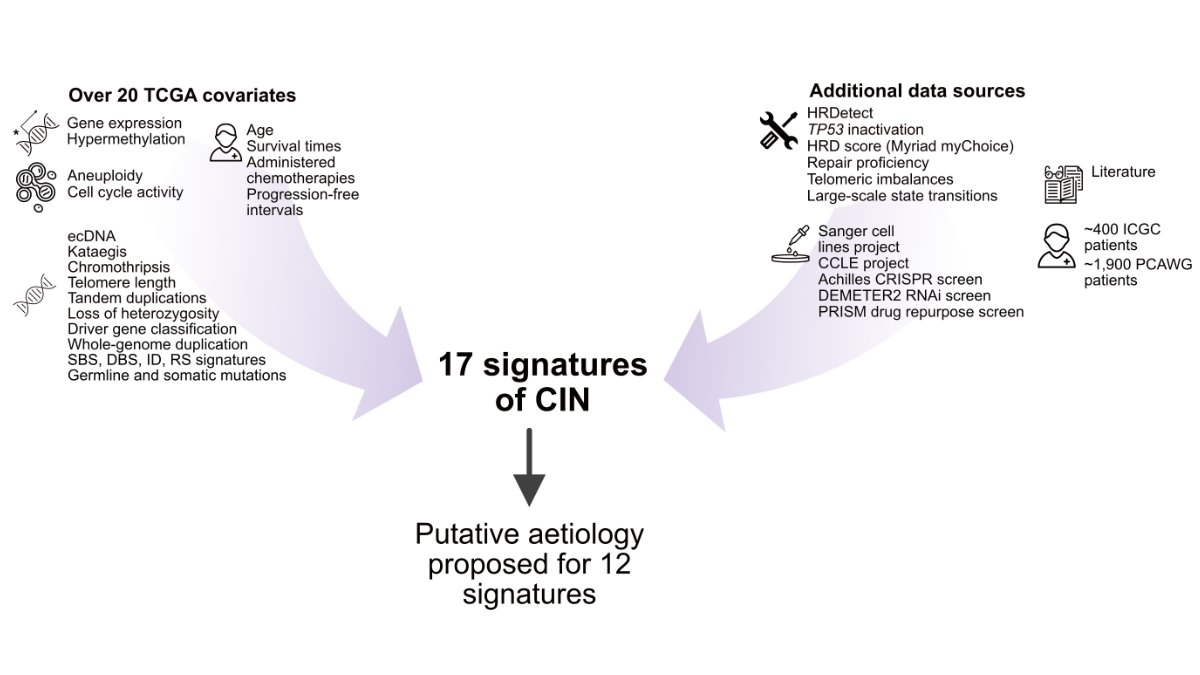
After climbing this huge mountain of interpretation, we got a view from the top of the mountain. 3 signals for mitotic errors; 3 signals for impaired homologous recombination; 4 signals for replication stress; 1 signal for impaired NHEJ; 1 signal for PI3K-mediated WGD (star rating = strength of evidence). 5 unknowns.
When we consider this perspective, we realize that many of the identified defective causal pathways contain drivers that are being treated. Can CIN signatures predict response to these therapies? Or even identify new drug targets? (You didn’t realize it, the first draft didn’t think of this at all)
To explore this, we combined the signatures of 297 cancer cell lines with genome-wide CRISPR, RNAi and drug screening data from CancerDepMap. We found that our signature can predict response to 44 targeted therapies and perturbation assessments of 49 CIN-related druggable genes.
Given these exciting in vitro results, we wondered…can we also predict patient response to one of the main treatments for CIN cancer: platinum-based chemotherapy? (This is also added by the reviewer diss)
HRD has previously been shown to predict sensitivity to platinum chemotherapy. But now, we have a way to break down the “HRD” phenotype using 3 IHR features.
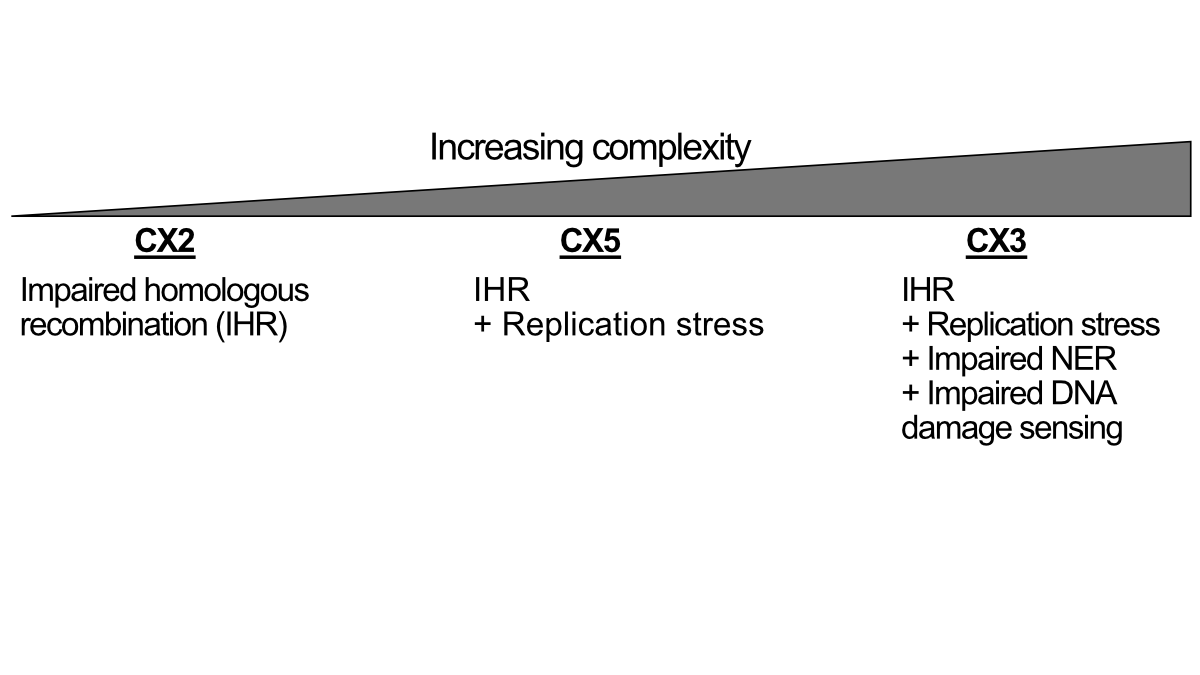
To test this, we focused on ovarian cancer for which platinum chemotherapy is routinely used. Using overall survival as a surrogate for sensitivity, we found that CX2 was not predictive, CX5 predicted resistance, and CX3 was sensitive! This is already great. But what if we combine them?
Based on the idea that germline BRCA1/2 mutants may have HR impairment but are not necessarily sensitive to platinum chemotherapy, we came up with a simple classification rule to exclude “background” CINs (CX2) from predictive CINs (CX3) . A simple rule, if CX3>CX2, then it is sensitive, otherwise it is resistant.
This simple classifier is powerful enough to dissect a patient population uniformly treated with platinum. This includes ovarian cancers with germline mutations in BRCA1, as well as other ovarian, esophageal, and breast cancer cohorts. Its performance is also comparable to more sophisticated methods.
What a journey, our signature provides a window into CIN and a framework to assess the extent, diversity and origin of pan-cancer chromosomal instability. (Sublimation theme, upper value)
To bring this technology to patients faster, we also founded TailorBio. The tech-bio startup has exclusive rights to translate this technology into a precision medicine platform for improving patient stratification and developing new targeted therapies.
The next research frontier is to study the dynamics of CIN. We are developing technologies to identify ongoing CINs using single-cell genomic profiling from early-stage lesions, KO cell lines, and drug-treated organs. You are welcome to contact us. (Introduce what you are doing, occupy the site and recruit people by the way)
The author of this article : Bear thinking about problems
Copyright notice : Unless otherwise stated, all articles on this blog are licensed under the Creative Commons Attribution-Non-Commercial-No Derivatives 4.0 International License Agreement (CC BY-NC-ND 4.0) .
This article is reprinted from: https://kaopubear.top/blog/2022-06-26-tcga-pancan-cin-signature/
This site is for inclusion only, and the copyright belongs to the original author.